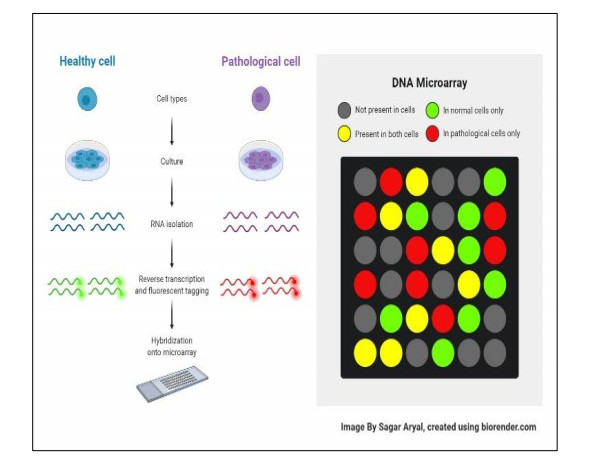
In this article, Autism Spectrum Disorder (ASD) is discussed, with an emphasis placed on the multidimensional nature of the disorder, which is anchored in genetic and neurological components. Identifying genes related to ASD is essential to comprehend the mechanisms that underlie the illness, yet the condition's complexity has impeded precise information in this field. In ASD research, the analysis of gene expression data helps choose and categorize significant genes. The study used microarray data to provide a novel approach that integrated gene selection techniques with deep learning models to improve the accuracy of ASD prediction. It offered a detailed comparative examination of gene selection approaches and deep learning architectures, including singular value decompositions (SVD), principal component analyses (PCA), and convolutional neural networks (CNNs). This paper combines gene selection methods (PCA and SVD) with deep learning models (CNN) to improve ASD prediction. Compared to more traditional approaches, the study revealed that its integrated methodology was more effective in improving the accuracy of ASD prediction results through experimentation. There was a difference in the accuracy between the PCA-CNN model, which achieved 94.33% with a loss of 0.4312, and the SVD-CNN model, which achieved 92.21% with a loss less than or equal to 0.3354. These discoveries help in the development of more accurate diagnostic and prognostic tools for ASD, which is a complicated neurodevelopmental disorder. Additionally, they provide insights into the molecular pathways that underlie ASD.
Citation: Mahmoud M. Abdelwahab, Khamis A. Al-Karawi, H. E. Semary. Integrating gene selection and deep learning for enhanced Autisms' disease prediction: a comparative study using microarray data[J]. AIMS Mathematics, 2024, 9(7): 17827-17846. doi: 10.3934/math.2024867
In this article, Autism Spectrum Disorder (ASD) is discussed, with an emphasis placed on the multidimensional nature of the disorder, which is anchored in genetic and neurological components. Identifying genes related to ASD is essential to comprehend the mechanisms that underlie the illness, yet the condition's complexity has impeded precise information in this field. In ASD research, the analysis of gene expression data helps choose and categorize significant genes. The study used microarray data to provide a novel approach that integrated gene selection techniques with deep learning models to improve the accuracy of ASD prediction. It offered a detailed comparative examination of gene selection approaches and deep learning architectures, including singular value decompositions (SVD), principal component analyses (PCA), and convolutional neural networks (CNNs). This paper combines gene selection methods (PCA and SVD) with deep learning models (CNN) to improve ASD prediction. Compared to more traditional approaches, the study revealed that its integrated methodology was more effective in improving the accuracy of ASD prediction results through experimentation. There was a difference in the accuracy between the PCA-CNN model, which achieved 94.33% with a loss of 0.4312, and the SVD-CNN model, which achieved 92.21% with a loss less than or equal to 0.3354. These discoveries help in the development of more accurate diagnostic and prognostic tools for ASD, which is a complicated neurodevelopmental disorder. Additionally, they provide insights into the molecular pathways that underlie ASD.

| [1] | W. H. Organization, Autism spectrum disorders, Regional Office for the Eastern Mediterranean, 2019. Available from: https://iris.who.int/handle/10665/364128. |
| [2] |
M. M. Abdelwahab, K. A. Al-Karawi, E. Hasanin, H. Semary, Autism spectrum disorder prediction in children using machine learning, J. Disability Res., 3 (2024), 1–9. https://doi.org/10.57197/JDR-2023-0064 doi: 10.57197/JDR-2023-0064

|
| [3] |
P. Hlavatá, T. Kašpárek, P. Linhartová, H. Ošlejšková, M. Bareš, Autism, impulsivity and inhibition a review of the literature, Basal Ganglia, 14 (2018), 44–53. https://doi.org/10.1016/j.baga.2018.10.002 doi: 10.1016/j.baga.2018.10.002
|
| [4] |
H. Semary, K. A. Al-Karawi, M. M. Abdelwahab, A. Elshabrawy, A review on internet of things (IoT)-related disabilities and their implications, J. Disability Res., 3 (2024), 1–16. https://doi.org/10.57197/JDR-2024-0012 doi: 10.57197/JDR-2024-0012
|
| [5] |
S. Heinsfeld, A. R. Franco, R. C. Craddock, A. Buchweitz, F. Meneguzzi, Identification of autism spectrum disorder using deep learning and the ABIDE dataset, NeuroImage: Clinical, 17 (2018), 16–23. https://doi.org/10.1016/j.nicl.2017.08.017 doi: 10.1016/j.nicl.2017.08.017
|
| [6] |
H. Semary, K. A. Al-Karawi, M. M. Abdelwahab, Using voice technologies to support disabled people, J. Disability Res., 3 (2024), 1–8. https://doi.org/10.57197/jdr-2023-0063 doi: 10.57197/jdr-2023-0063
|
| [7] | L. Franz, K. Adewumi, N. Chambers, M. Viljoen, J. N. Baumgartner, P. J. De Vries, Providing early detection and early intervention for autism spectrum disorder in South Africa: stakeholder perspectives from the Western Cape province, J. Child Adolesc. Mental Health, 30 (2018), 149–165. |
| [8] |
K. A. Al-karawi, Real-time adaptive training for forensic speaker verification in reverberation conditions, Int. J. Speech Technol., 26 (2023), 1079–1089. https://doi.org/10.1007/s10772-023-10074-5 doi: 10.1007/s10772-023-10074-5
|
| [9] |
M. Pagnozzi, E. Conti, S. Calderoni, J. Fripp, S. E. Rose, A systematic review of structural MRI biomarkers in autism spectrum disorder: a machine learning perspective, Int. J. Dev. Neurosci., 71 (2018), 68–82. https://doi.org/10.1016/j.ijdevneu.2018.08.010 doi: 10.1016/j.ijdevneu.2018.08.010
|
| [10] | S. Alenizi, K. A. Al-karawi, Cloud computing adoption-based digital open government services: challenges and barriers, In: X. S. Yang, S. Sherratt, N. Dey, A. Joshi, Proceedings of Sixth International Congress on Information and Communication Technology, Singapore: Springer, 216 (2022), 149–160. https://doi.org/10.1007/978-981-16-1781-2_15 |
| [11] |
F. Thabtah, Machine learning in autistic spectrum disorder behavioral research: a review and ways forward, Inform. Health Soc. Care, 44 (2019), 278–297. https://doi.org/10.1080/17538157.2017.1399132 doi: 10.1080/17538157.2017.1399132
|
| [12] |
K. A. Al-Karawi, D. Y. Mohammed, Early reflection detection using autocorrelation to improve robustness of speaker verification in reverberant conditions, Int. J. Speech Technol., 22 (2019), 1077–1084. https://doi.org/10.1007/s10772-019-09648-z doi: 10.1007/s10772-019-09648-z
|
| [13] |
U. Frith, F. Happé, Autism spectrum disorder, Curr. Biol., 15 (2005), R786–R790. https://doi.org/10.1016/j.cub.2005.09.033 doi: 10.1016/j.cub.2005.09.033
|
| [14] | K. A. Al-Karawi, B. Al-Bayati, The effects of distance and reverberation time on speaker recognition performance, Int. J. Inform. Technol., 2024. https://doi.org/10.1007/s41870-024-01789-y |
| [15] |
H. K. Tripathy, P. K. Mallick, S. Mishra, Application and evaluation of classification model to detect autistic spectrum disorders in children, Int. J. Comput. Appl. Technol., 65 (2021), 368–377. https://doi.org/10.1504/IJCAT.2021.117286 doi: 10.1504/IJCAT.2021.117286
|
| [16] |
K. A. Al-Karawi, D. Y. Mohammed, Improving short utterance speaker verification by combining MFCC and Entrocy in Noisy conditions, Multimedia Tools Appl., 80 (2021), 22231–22249. https://doi.org/10.1007/s11042-021-10767-6 doi: 10.1007/s11042-021-10767-6
|
| [17] | K. S. Omar, P. Mondal, N. S. Khan, M. R. K. Rizvi, M. N. Islam, A machine learning approach to predict autism spectrum disorder, 2019 International conference on electrical, computer and communication engineering (ECCE), 2019. https://doi.org/10.1109/ECACE.2019.8679454 |
| [18] | S. Alenizi, K. A. Al-Karawi, Effective biometric technology used with big data, In: X. S. Yang, S. Sherratt, N. Dey, A. Joshi, Proceedings of Seventh International Congress on Information and Communication Technology, Singapore: Springer, 464 (2023), 239–250. https://doi.org/10.1007/978-981-19-2394-4_22 |
| [19] |
J. A. Bastiaansen, M. Thioux, L. Nanetti, C. van der Gaag, C. Ketelaars, R. Minderaa, et al., Age-related increase in inferior frontal gyrus activity and social functioning in autism spectrum disorder, Biol. Psychiatry, 69 (2011), 832–838. https://doi.org/10.1016/j.biopsych.2010.11.007 doi: 10.1016/j.biopsych.2010.11.007
|
| [20] | S. Alenizi, K. A. Al-Karawi, Internet of things (IoT) adoption: challenges and barriers, In: X. S. Yang, S. Sherratt, N. Dey, A. Joshi, Proceedings of Seventh International Congress on Information and Communication Technology, Singapore: Springer, 464 (2023), 217–229. https://doi.org/10.1007/978-981-19-2394-4_20 |
| [21] | S. Alenizi, K. A. Al-karawi, Machine learning approach for diabetes prediction, In: X. S. Yang, S. Sherratt, N. Dey, A. Joshi, Proceedings of Eighth International Congress on Information and Communication Technology, ICICT 2023, Lecture Notes in Networks and Systems, Singapore: Springer, 695 (2023), 745–756. https://doi.org/10.30534/ijiscs/2019/13822019 |
| [22] | G. Suhas, N. Naveen, M. Nagabanu, N. Kumar, Premature identification of autism spectrum disorder using machine learning techniques, Adv. Innovations Comput. Program. Languages, 3 (2021), 1–10. |
| [23] |
K. A. Al-Karawi, Face mask effects on speaker verification performance in the presence of noise, Multimedia Tools Appl., 83 (2023), 4811–4824. https://doi.org/10.1007/s11042-023-15824-w doi: 10.1007/s11042-023-15824-w
|
| [24] | R. Vaishali, R. Sasikala, A machine learning based approach to classify autism with optimum behaviour sets, Int. J. Eng. Technol., 7 (2018), 18. |
| [25] |
M. S. Othman, S. R. Kumaran, L. M. Yusuf, Gene selection using hybrid multi-objective cuckoo search algorithm with evolutionary operators for cancer microarray data, IEEE Access, 8 (2020), 186348–186361. https://doi.org/10.1109/ACCESS.2020.3029890 doi: 10.1109/ACCESS.2020.3029890
|
| [26] |
W. Zhongxin, S. Gang, Z. Jing, Z. Jia, Feature selection algorithm based on mutual information and lasso for microarray data, Open Biotechnol. J., 10 (2016), 278–286. https://doi.org/10.2174/1874070701610010278 doi: 10.2174/1874070701610010278
|
| [27] |
J. Zahoor, K. Zafar, Classification of microarray gene expression data using an infiltration tactics optimization (ITO) algorithm, Genes, 11 (2020), 819. https://doi.org/10.3390/genes11070819 doi: 10.3390/genes11070819
|
| [28] |
K. A. Al-Karawi, S. T. Ahmed, Model selection toward robustness speaker verification in reverberant conditions, Multimedia Tools Appl., 80 (2021), 36549–36566. https://doi.org/10.1007/s11042-021-11356-3 doi: 10.1007/s11042-021-11356-3
|
| [29] | M. Babu, K. Sarkar, A comparative study of gene selection methods for cancer classification using microarray data, 2016 Second International Conference on Research in Computational Intelligence and Communication Networks (ICRCICN), 2016,204–211. https://doi.org/10.1109/ICRCICN.2016.7813657 |
| [30] |
K. A. Al-Karawi, D. Y. Mohammed, Using combined features to improve speaker verification in the face of limited reverberant data, Int. J. Speech Technol., 26 (2023), 789–799. https://doi.org/10.1007/s10772-023-10048-7 doi: 10.1007/s10772-023-10048-7
|
| [31] | L. Yu, H. Liu, Feature selection for high-dimensional data: a fast correlation-based filter solution, Proceedings of the 20th international conference on machine learning (ICML-03), 2003,856–863. |
| [32] | D. H. Lim, Principal component analysis using singular value decomposition of microarray data, Int. J. Math. Comput. Phys. Quantum Eng., 7 (2013), 1390–1392. |
| [33] | M. Dufva, Introduction to microarray technology, In: M. Dufva, DNA microarrays for biomedical research, Methods and Protocols, 529 (2009), 1–22. https://doi.org/10.1007/978-1-59745-538-1_1 |
| [34] | U. R. Müller, D. V. Nicolau, Microarray technology and its applications, Springer, 2005. https://doi.org/10.1007/b137842 |
| [35] |
R. Govindarajan, J. Duraiyan, K. Kaliyappan, M. Palanisamy, Microarray and its applications, J. Pharm. Bioallied Sci., 4 (2012), S310–S312. https://doi.org/10.4103/0975-7406.100283 doi: 10.4103/0975-7406.100283
|
| [36] |
R. Kothapalli, S. J. Yoder, S. Mane, T. P. Loughran, Microarray results: how accurate are they, BMC Bioinf., 3 (2002), 22. https://doi.org/10.1186/1471-2105-3-22 doi: 10.1186/1471-2105-3-22
|
| [37] |
D. H. Blohm, A. Guiseppi-Elie, New developments in microarray technology, Curr. Opin. Biotech., 12 (2001), 41–47. https://doi.org/10.1016/S0958-1669(00)00175-0 doi: 10.1016/S0958-1669(00)00175-0
|
| [38] |
M. M. Abdelwahab, K. A. Al-Karawi, H. E. Semary, Deep learning-based prediction of Alzheimer's disease using microarray gene expression data, Biomedicines, 11 (2023), 3304. https://doi.org/10.3390/biomedicines11123304 doi: 10.3390/biomedicines11123304
|
| [39] |
S. Abrahams, D. E. Arking, D. B. Campbell, H. C. Mefford, E. M. Morrow, L. A. Weiss, et al., SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs), Mol. Autism, 4 (2013), 36. https://doi.org/10.1186/2040-2392-4-36 doi: 10.1186/2040-2392-4-36
|
| [40] |
C. Yang, J. Li, Q. Wu, X. Yang, A. Y. Huang, J. Zhang, et al., AutismKB 2.0: a knowledgebase for the genetic evidence of autism spectrum disorder, Database, 2018 (2018), bay106. https://doi.org/10.1093/database/bay106 doi: 10.1093/database/bay106
|
| [41] | L. Kolberg, U. Raudvere, I. Kuzmin, J. Vilo, H. Peterson, gprofiler2--an R package for gene list functional enrichment analysis and namespace conversion toolset g: Profiler, F1000Res., 9 (2020), ELIXIR-709. https://doi.org/10.12688/f1000research.24956.2 |
| [42] | H. Ahmed, H. Soliman, M. Elmogy, Early detection of Alzheimer's disease based on single nucleotide polymorphisms (SNPs) analysis and machine learning techniques, 2020 International Conference on Data Analytics for Business and Industry: Way Towards a Sustainable Economy (ICDABI), 2020, 1–6. https://doi.org/10.1109/ICDABI51230.2020.9325640 |
| [43] |
M. Lenz, F. J. Müller, M. Zenke, A. Schuppert, Principal components analysis and the reported low intrinsic dimensionality of gene expression microarray data, Sci. Rep., 6 (2016), 25696. https://doi.org/10.1038/srep25696 doi: 10.1038/srep25696
|
| [44] | N. Parveen, H. H. Inbarani, E. N. S. Kumar, Performance analysis of unsupervised feature selection methods, 2012 International Conference on Computing, Communication and Applications, 2012, 1–7. https://doi.org/10.1109/ICCCA.2012.6179181 |
| [45] |
Y. Zhang, J. M. Gorriz, Z. Dong, Deep learning in medical image analysis, J. Imaging, 7 (2021), 74. https://doi.org/10.3390/jimaging7040074 doi: 10.3390/jimaging7040074
|
| [46] |
M. Mostavi, Y. Chiu, Y. Huang, Y. Chen, Convolutional neural network models for cancer type prediction based on gene expression, BMC Med. Genomics, 13 (2020), 44. https://doi.org/10.1186/s12920-020-0677-2 doi: 10.1186/s12920-020-0677-2
|
| [47] |
S. Kiranyaz, O. Avci, O. Abdeljaber, T. Ince, M. Gabbouj, D. Inman, 1D convolutional neural networks and applications: a survey, Mech. Syst. Signal Process., 151 (2021), 107398. https://doi.org/10.1016/j.ymssp.2020.107398 doi: 10.1016/j.ymssp.2020.107398
|
| [48] |
T. Ragunthar, S. Selvakumar, Classification of gene expression data with optimized feature selection, Int. J. Recent Technol. Eng., 8 (2019), 4763–4769. https://doi.org/10.35940/ijrte.b1845.078219 doi: 10.35940/ijrte.b1845.078219
|
| [49] |
J. R. Vergara, P. A. Estévez, A review of feature selection methods based on mutual information, Neural Comput. Appl., 24 (2014), 175–186. https://doi.org/10.1007/s00521-013-1368-0 doi: 10.1007/s00521-013-1368-0
|




Figures(5) / Tables(3)

Mahmoud M. Abdelwahab, Khamis A. Al-Karawi, H. E. Semary. Integrating gene selection and deep learning for enhanced Autisms' disease prediction: a comparative study using microarray data[J]. AIMS Mathematics, 2024, 9(7): 17827-17846. doi: 10.3934/math.2024867







 DownLoad:
DownLoad: