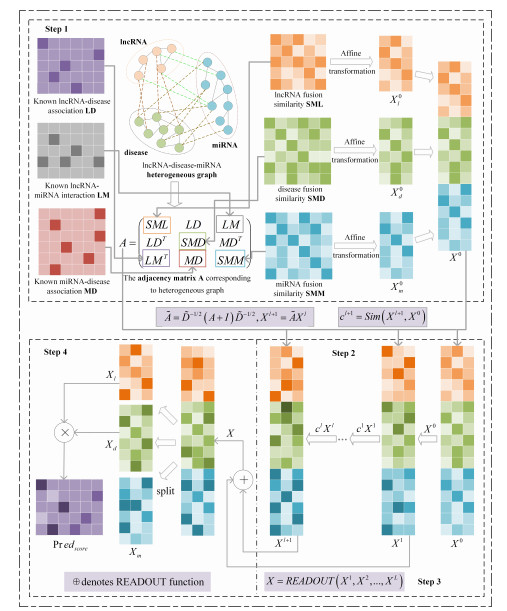
Long non-coding RNA (lncRNA) is considered to be a crucial regulator involved in various human biological processes, including the regulation of tumor immune checkpoint proteins. It has great potential as both a cancer biomolecular biomarker and therapeutic target. Nevertheless, conventional biological experimental techniques are both resource-intensive and laborious, making it essential to develop an accurate and efficient computational method to facilitate the discovery of potential links between lncRNAs and diseases. In this study, we proposed HRGCNLDA, a computational approach utilizing hierarchical refinement of graph convolutional neural networks for forecasting lncRNA-disease potential associations. This approach effectively addresses the over-smoothing problem that arises from stacking multiple layers of graph convolutional neural networks. Specifically, HRGCNLDA enhances the layer representation during message propagation and node updates, thereby amplifying the contribution of hidden layers that resemble the ego layer while reducing discrepancies. The results of the experiments showed that HRGCNLDA achieved the highest AUC-ROC (area under the receiver operating characteristic curve, AUC for short) and AUC-PR (area under the precision versus recall curve, AUPR for short) values compared to other methods. Finally, to further demonstrate the reliability and efficacy of our approach, we performed case studies on the case of three prevalent human diseases, namely, breast cancer, lung cancer and gastric cancer.
Citation: Li Peng, Yujie Yang, Cheng Yang, Zejun Li, Ngai Cheong. HRGCNLDA: Forecasting of lncRNA-disease association based on hierarchical refinement graph convolutional neural network[J]. Mathematical Biosciences and Engineering, 2024, 21(4): 4814-4834. doi: 10.3934/mbe.2024212
Long non-coding RNA (lncRNA) is considered to be a crucial regulator involved in various human biological processes, including the regulation of tumor immune checkpoint proteins. It has great potential as both a cancer biomolecular biomarker and therapeutic target. Nevertheless, conventional biological experimental techniques are both resource-intensive and laborious, making it essential to develop an accurate and efficient computational method to facilitate the discovery of potential links between lncRNAs and diseases. In this study, we proposed HRGCNLDA, a computational approach utilizing hierarchical refinement of graph convolutional neural networks for forecasting lncRNA-disease potential associations. This approach effectively addresses the over-smoothing problem that arises from stacking multiple layers of graph convolutional neural networks. Specifically, HRGCNLDA enhances the layer representation during message propagation and node updates, thereby amplifying the contribution of hidden layers that resemble the ego layer while reducing discrepancies. The results of the experiments showed that HRGCNLDA achieved the highest AUC-ROC (area under the receiver operating characteristic curve, AUC for short) and AUC-PR (area under the precision versus recall curve, AUPR for short) values compared to other methods. Finally, to further demonstrate the reliability and efficacy of our approach, we performed case studies on the case of three prevalent human diseases, namely, breast cancer, lung cancer and gastric cancer.

| [1] |
Y. J. Chi, D. Wang, J. P. Wang, W. D. Yu, J. C. Yang, Long non-coding rna in the pathogenesis of cancers, Cells, 8 (2019), 1015. https://doi.org/10.3390/cells8091015 doi: 10.3390/cells8091015

|
| [2] |
S. Djebali, C. A. Davis, A. Merkel, A. Dobin, T. Lassmann, A. Mortazavi, et al., Landscape of transcription in human cells, Nature, 489 (2012), 101–108. https://doi.org/10.1038/nature11233 doi: 10.1038/nature11233
|
| [3] |
A. T. Willingham, A. P. Orth, S. Batalov, E. C. Peters, B. G. Wen, P. Aza-Blanc, et al., A strategy for probing the function of noncoding rnas finds a repressor of nfat, Science, 309 (2005), 1570–1573. https://doi.org/10.1126/science.1115901 doi: 10.1126/science.1115901
|
| [4] |
C. Xing, S. G. Sun, Z. Q. Yue, F. Bai, Role of lncrna lucat1 in cancer, Biomed. Pharmacother., 134 (2021), 111158. https://doi.org/10.1016/j.biopha.2020.111158 doi: 10.1016/j.biopha.2020.111158
|
| [5] |
L. Peng, M. Peng, B. Liao, G. H. Huang, W. B. Li, D. F. Xie, The advances and challenges of deep learning application in biological big data processing, Curr. Bioinf., 13 (2018), 352–359. https://doi.org/10.1163/9789004392533_041 doi: 10.1163/9789004392533_041
|
| [6] |
R. H. Wang, Y. Jiang, J. R. Jin, C. L. Yin, H. Q. Yu, F. S. Wang, et al., Deepbio: an automated and interpretable deep-learning platform for high-throughput biological sequence prediction, functional annotation and visualization analysis, Nucleic Acids Res., 51 (2023), 3017–3029. https://doi.org/10.1093/nar/gkad055 doi: 10.1093/nar/gkad055
|
| [7] |
L. H. Peng, J. W. Tan, W. Xiong, L. Zhang, Z. Wang, R. Y. Yuan, et al., Deciphering ligand-receptor-mediated intercellular communication based on ensemble deep learning and the joint scoring strategy from single-cell transcriptomic data, Comput. Biol. Med., 163 (2023), 107137. https://doi.org/10.1016/j.compbiomed.2023.107137 doi: 10.1016/j.compbiomed.2023.107137
|
| [8] |
W. Liu, Y. Yang, X. Lu, X. Z. Fu, R. Q. Sun, L. Yang, et al., Nsrgrn: a network structure refinement method for gene regulatory network inference, Briefings Bioinf., 24 (2023), bbad129. https://doi.org/10.1093/bib/bbad129 doi: 10.1093/bib/bbad129
|
| [9] |
J. C. Wang, Y. J. Chen, Q. Zou, Inferring gene regulatory network from single-cell transcriptomes with graph autoencoder model, PLos Genet., 19 (2023), e1010942. https://doi.org/10.1371/journal.pgen.1010942 doi: 10.1371/journal.pgen.1010942
|
| [10] |
L. Peng, C. Yang, L. Huang, X. Chen, X. Z. Fu, W. Liu, Rnmflp: predicting circrna-disease associations based on robust nonnegative matrix factorization and label propagation, Briefings Bioinf., 23 (2022), bbac155. https://doi.org/10.1093/bib/bbac155 doi: 10.1093/bib/bbac155
|
| [11] |
W. Liu, T. T. Tang, X. Lu, X. Z. Fu, Y. Yang, L. Peng, Mpclcda: predicting circrna-disease associations by using automatically selected meta-path and contrastive learning, Briefings Bioinf., 24 (2023), bbad227. https://doi.org/10.1093/bib/bbad227 doi: 10.1093/bib/bbad227
|
| [12] |
L. L. Zhuo, S. Y. Pan, J. Li, X. Z. Fu, Predicting mirna-lncrna interactions on plant datasets based on bipartite network embedding method, Methods, 207 (2022), 97–102. https://doi.org/10.1016/j.ymeth.2022.09.002 doi: 10.1016/j.ymeth.2022.09.002
|
| [13] |
Z. C. Zhou, Z. Y. Du, J. H. Wei, L. L. Zhuo, S. Y. Pan, X. Z. Fu, et al., Mham-npi: Predicting ncrna-protein interactions based on multi-head attention mechanism, Comput. Biol. Med., 163 (2023), 107143. https://doi.org/10.1016/j.compbiomed.2023.107143 doi: 10.1016/j.compbiomed.2023.107143
|
| [14] |
X. Chen, L. Wang, J. Qu, N. N. Guan, J. Q. Li, Predicting mirna-disease association based on inductive matrix completion, Bioinformatics, 34 (2018), 4256–4265. https://doi.org/10.1093/bioinformatics/bty503 doi: 10.1093/bioinformatics/bty503
|
| [15] |
X. Chen, D. Xie, L. Wang, Q. Zhao, Z. H. You, H. Liu, Bnpmda: Bipartite network projection for mirna-disease association prediction, Bioinformatics, 34 (2018), 3178–3186. https://doi.org/10.1093/bioinformatics/bty333 doi: 10.1093/bioinformatics/bty333
|
| [16] |
L. Huang, L. Zhang, X. Chen, Updated review of advances in micrornas and complex diseases: towards systematic evaluation of computational models, Briefings Bioinf., 23 (2022), bbac407. https://doi.org/10.1093/bib/bbac407 doi: 10.1093/bib/bbac407
|
| [17] |
C. C. Wang, C. C. Zhu, X. Chen, Ensemble of kernel ridge regression-based small molecule-mirna association prediction in human disease, Briefings Bioinf., 23 (2022), bbab431. https://doi.org/10.1093/bib/bbab431 doi: 10.1093/bib/bbab431
|
| [18] |
Z. J. Li, Y. X. Zhang, Y. Bai, X. H. Xie, L. J. Zeng, Imc-mda: Prediction of mirna-disease association based on induction matrix completion, Math. Biosci. Eng., 20 (2023), 10659–10674. https://doi.org/10.3934/mbe.2023471 doi: 10.3934/mbe.2023471
|
| [19] |
Q. Qu, X. Chen, B. Ning, X. Zhang, H. Nie, L. Zeng, et al., Prediction of mirna-disease associations by neural network-based deep matrix factorization, Methods, 212 (2023), 1–9. https://doi.org/10.1016/j.ymeth.2023.02.003 doi: 10.1016/j.ymeth.2023.02.003
|
| [20] |
L. Zhang, C. C. Wang, X. Chen, Predicting drug-target binding affinity through molecule representation block based on multi-head attention and skip connection, Briefings Bioinf., 23 (2022), bbac468. https://doi.org/10.1093/bib/bbac468 doi: 10.1093/bib/bbac468
|
| [21] |
L. Katusiime, Covid-19 and the effect of central bank intervention on exchange rate volatility in developing countries: The case of uganda, National Accounting Rev., 5 (2023), 23–37. https://doi.org/10.3934/NAR.2023002 doi: 10.3934/NAR.2023002
|
| [22] |
L. Grassini, Statistical features and economic impact of Covid-19, National Accounting Rev., 5 (2023), 38–40. https://doi.org/10.3934/NAR.2023003 doi: 10.3934/NAR.2023003
|
| [23] |
Z. Y. Bao, Z. Yang, Z. Huang, Y. R. Zhou, Q. H. Cui, D. Dong, Lncrnadisease 2.0: an updated database of long non-coding rna-associated diseases, Nucleic Acids Res., 47 (2019), D1034–D1037. https://doi.org/10.1093/nar/gky905 doi: 10.1093/nar/gky905
|
| [24] |
S. W. Ning, J. Z. Zhang, P. Wang, H. Zhi, J. J. Wang, Y. Liu, et al., Lnc2cancer: a manually curated database of experimentally supported lncrnas associated with various human cancers, Nucleic Acids Res., 44 (2016), D980–D985. https://doi.org/10.1093/nar/gkv1094 doi: 10.1093/nar/gkv1094
|
| [25] |
X. Chen, L. Huang, Computational model for disease research, Briefings Bioinf., 24 (2023), bbac615. https://doi.org/10.1093/bib/bbac615 doi: 10.1093/bib/bbac615
|
| [26] |
K. Albitar, K. Hussainey, Sustainability, environmental responsibility and innovation, Green Finance, 5 (2023), 85–88. https://doi.org/10.3934/GF.2023004 doi: 10.3934/GF.2023004
|
| [27] |
G. Desalegn, Insuring a greener future: How green insurance drives investment in sustainable projects in developing countries, Green Finance, 5 (2023), 195–210. https://doi.org/10.3934/GF.2023008 doi: 10.3934/GF.2023008
|
| [28] |
Y. Liang, Z. H. Zhang, N. N. Liu, Y. N. Wu, C. L. Gu, Y. L. Wang, Magcnse: predicting lncrna-disease associations using multi-view attention graph convolutional network and stacking ensemble model, BMC Bioinf., 23 (2022). https://doi.org/10.1186/s12859-022-04715-w doi: 10.1186/s12859-022-04715-w
|
| [29] |
Y. Kim, M. Lee, Deep learning approaches for lncrna-mediated mechanisms: A comprehensive review of recent developments, Int. J. Mol. Sci., 24 (2023), 10299. https://doi.org/10.3390/ijms241210299 doi: 10.3390/ijms241210299
|
| [30] |
Z. Q. Zhang, J. L. Xu, Y. N. Wu, N. N. Liu, Y. L. Wang, Y. Liang, Capsnet-lda: predicting lncrna-disease associations using attention mechanism and capsule network based on multi-view data, Briefings Bioinf., 24 (2022), bbac531. https://doi.org/10.1093/bib/bbac531 doi: 10.1093/bib/bbac531
|
| [31] |
N. Dwarika, The risk-return relationship and volatility feedback in south africa: a comparative analysis of the parametric and nonparametric bayesian approach, Quant. Finance Econ., 7 (2023), 119–146. https://doi.org/10.3934/QFE.2023007 doi: 10.3934/QFE.2023007
|
| [32] |
N. Dwarika, Asset pricing models in south africa: A comparative of regression analysis and the bayesian approach, Data Sci. Finance Econ., 3 (2023), 55–75. https://doi.org/10.3934/DSFE.2023004 doi: 10.3934/DSFE.2023004
|
| [33] |
Y. Q. Lin, X. J. Chen, H. Y. Lan, Analysis and prediction of american economy under different government policy based on stepwise regression and support vector machine modelling, Data Sci. Finance Econ., 3 (2023), 1–13. https://doi.org/10.3934/DSFE.2023001 doi: 10.3934/DSFE.2023001
|
| [34] |
N. Sheng, L. Huang, Y. T. Lu, H. Wang, L. L. Yang, L. Gao, et al., Data resources and computational methods for lncrna-disease association prediction, Comput. Biol. Med., 153 (2023), 106527. https://doi.org/10.1016/j.compbiomed.2022.106527 doi: 10.1016/j.compbiomed.2022.106527
|
| [35] |
J. H. Wei, L. L. Zhuo, S. Y. Pan, X. Z. Lian, X. J. Yao, X. Z. Fu, Headtailtransfer: An efficient sampling method to improve the performance of graph neural network method in predicting sparse ncrna-protein interactions, Comput. Biol. Med., 157 (2023), 106783. https://doi.org/10.1016/j.compbiomed.2023.106783 doi: 10.1016/j.compbiomed.2023.106783
|
| [36] |
P. Xuan, S. X. Pan, T. G. Zhang, Y. Liu, H. Sun, Graph convolutional network and convolutional neural network based method for predicting lncrna-disease associations, Cells, 8 (2019), 1012. https://doi.org/10.3390/cells8091012 doi: 10.3390/cells8091012
|
| [37] |
M. F. Leung, A. Jawaid, S. W. Ip, C. H. Kwok, S. Yan, A portfolio recommendation system based on machine learning and big data analytics, Data Sci. Finance Econ., 3 (2023), 152–165. https://doi.org/10.3934/DSFE.2023009 doi: 10.3934/DSFE.2023009
|
| [38] |
Q. W. Wu, J. F. Xia, J. C. Ni, C. H. Zheng, Gaerf: predicting lncrna-disease associations by graph auto-encoder and random forest, Briefings Bioinf., 22 (2021), bbaa391. https://doi.org/10.1093/bib/bbaa391 doi: 10.1093/bib/bbaa391
|
| [39] |
N. Sheng, L. Huang, Y. Wang, J. Zhao, P. Xuan, L. Gao, et al., Multi-channel graph attention autoencoders for disease-related lncrnas prediction, Briefings Bioinf., 23 (2022), bbab604. https://doi.org/10.1093/bib/bbab604 doi: 10.1093/bib/bbab604
|
| [40] |
L. Peng, C. Yang, Y. F. Chen, W. Liu, Predicting circrna-disease associations via feature convolution learning with heterogeneous graph attention network, IEEE J. Biomed. Health. Inf., 27 (2023), 3072–3082. https://doi.org/10.1109/JBHI.2023.3260863. doi: 10.1109/JBHI.2023.3260863
|
| [41] |
X. Liu, C. Z. Song, F. Huang, H. T. Fu, W. J. Xiao, W. Zhang, Graphcdr: a graph neural network method with contrastive learning for cancer drug response prediction, Briefings Bioinf., 23 (2022), bbab457. https://doi.org/10.1093/bib/bbab457 doi: 10.1093/bib/bbab457
|
| [42] |
G. Y. Fu, J. Wang, C. Domeniconi, G. X. Yu, Matrix factorization-based data fusion for the prediction of lncrna–disease associations, Bioinformatics, 34 (2018), 1529–1537. https://doi.org/10.1093/bioinformatics/btx794 doi: 10.1093/bioinformatics/btx794
|
| [43] | Z. Y. Lu, K. B. Cohen, L. Hunter, Generif quality assurance as summary revision, in Biocomputing 2007, World Scientific, (2007), 269–280. https://doi.org/10.1142/9789812772435_0026 |
| [44] |
J. H. Li, S. Liu, H. Zhou, L. H. Qu, J. H. Yang, starBase v2. 0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data, Nucleic Acids Res., 42 (2014), D92–D97. https://doi.org/10.1093/nar/gkt1248 doi: 10.1093/nar/gkt1248
|
| [45] |
W. Lan, Y. Dong, Q. F. Chen, R. Q. Zheng, J. Liu, Y. Pan, et al., Kgancda: predicting circrna-disease associations based on knowledge graph attention network, Briefings Bioinf., 23 (2022), bbab494. https://doi.org/10.1093/bib/bbab494 doi: 10.1093/bib/bbab494
|
| [46] |
Z. H. Guo, Z. H. You, D. S. Huang, H. C. Yi, Z. H. Chen, Y. B. Wang, A learning based framework for diverse biomolecule relationship prediction in molecular association network, Commun. Biol., 3 (2020). https://doi.org/10.1038/s42003-020-0858-8 doi: 10.1038/s42003-020-0858-8
|
| [47] |
D. Wang, J. Wang, M. Lu, F. Song, Q. H. Cui, Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases, Bioinformatics, 26 (2010), 1644–1650. https://doi.org/10.1093/bioinformatics/btq241 doi: 10.1093/bioinformatics/btq241
|
| [48] |
X. Chen, Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA, Sci. Rep., 5 (2015), 13186. https://doi.org/10.1038/srep13186 doi: 10.1038/srep13186
|
| [49] |
X. Chen, G. Y. Yan, Novel human lncrna-disease association inference based on lncrna expression profiles, Bioinformatics, 29 (2013), 2617–2624. https://doi.org/10.1093/bioinformatics/btt426 doi: 10.1093/bioinformatics/btt426
|
| [50] |
D. Anderson, U. Ulrych, Accelerated american option pricing with deep neural networks, Quant. Finance Econ., 7 (2023), 207–228. https://doi.org/10.3934/QFE.2023011 doi: 10.3934/QFE.2023011
|
| [51] | T. N. Kipf, M. Welling, Semi-supervised classification with graph convolutional networks, preprint, arXiv: 1609.02907. https://doi.org/10.48550/arXiv.1609.02907 |
| [52] |
L. Peng, Y. Tu, L. Huang, Y. Li, X. Z. Fu, X. Chen, Daestb: inferring associations of small molecule–mirna via a scalable tree boosting model based on deep autoencoder, Briefings Bioinf., 23 (2022), bbac478. https://doi.org/10.1093/bib/bbac478 doi: 10.1093/bib/bbac478
|
| [53] |
Z. Y. Chu, S. C. Liu, W. Zhang, Hierarchical graph representation learning for the prediction of drug-target binding affinity, Inf. Sci., 613 (2022), 507–523. https://doi.org/10.1016/j.ins.2022.09.043 doi: 10.1016/j.ins.2022.09.043
|
| [54] | M. Chen, Z. W. Wei, Z. F. Huang, B. L. Ding, Y. L. Li, Simple and deep graph convolutional networks, in Proceedings of the 37th International Conference on Machine Learning, PMLR, (2020), 1725–1735. |
| [55] |
X. Chen, Katzlda: Katz measure for the lncrna-disease association prediction, Sci. Rep., 5 (2015), 16840. https://doi.org/10.1038/srep16840 doi: 10.1038/srep16840
|
| [56] |
C. Q. Lu, M. Y. Yang, F. Luo, F. X. Wu, M. Li, Y. Pan, et al., Prediction of lncrna–disease associations based on inductive matrix completion, Bioinformatics, 34 (2018), 3357–3364. https://doi.org/10.1093/bioinformatics/bty327 doi: 10.1093/bioinformatics/bty327
|
| [57] |
X. M. Wu, W. Lan, Q. F. Chen, Y. Dong, J. Liu, W. Peng, Inferring LncRNA-disease associations based on graph autoencoder matrix completion, Comput. Biol. Chem., 87 (2020), 107282. https://doi.org/10.1016/j.compbiolchem.2020.107282 doi: 10.1016/j.compbiolchem.2020.107282
|
| [58] |
M. Zeng, C. Q. Lu, Z. H. Fei, F. X. Wu, Y. H. Li, J. X. Wang, et al., Dmflda: a deep learning framework for predicting lncrna–disease associations, IEEE/ACM Trans. Comput. Biol. Bioinf., 18 (2020), 2353–2363. https://doi.org/10.1109/TCBB.2020.2983958. doi: 10.1109/TCBB.2020.2983958
|
| [59] |
R. Zhu, Y. Wang, J. X. Liu, L. Y. Dai, Ipcarf: improving lncrna-disease association prediction using incremental principal component analysis feature selection and a random forest classifier, BMC Bioinf., 22 (2021). https://doi.org/10.1186/s12859-021-04104-9 doi: 10.1186/s12859-021-04104-9
|
| [60] |
Y. S. Sun, Z. Zhao, Z. N. Yang, F. Xu, H. J. Lu, Z. Y. Zhu, et al., Risk factors and preventions of breast cancer, Int. J. Biol. Sci., 13 (2017), 1387–1397. https://doi.org/10.7150/ijbs.21635 doi: 10.7150/ijbs.21635
|
| [61] |
H. Jin, W. Du, W. T. Huang, J. J. Yan, Q. Tang, Y. B. Chen, et al., lncRNA and breast cancer: Progress from identifying mechanisms to challenges and opportunities of clinical treatment, Mol. Ther.–Nucleic Acids, 25 (2021), 613–637. https://doi.org/10.1016/j.omtn.2021.08.005 doi: 10.1016/j.omtn.2021.08.005
|
| [62] |
J. J. Xu, M. S. Hu, Y. Gao, Y. S. Wang, X. N. Yuan, Y. Yang, et al., Lncrna mir17hg suppresses breast cancer proliferation and migration as cerna to target fam135a by sponging mir-454-3p, Mol. Biotechnol., 65 (2023), 2071–2085. https://doi.org/10.1007/s12033-023-00706-1 doi: 10.1007/s12033-023-00706-1
|
| [63] | K. X. Lou, Z. H. Li, P. Wang, Z. Liu, Y. Chen, X. L. Wang, et al., Long non-coding rna bancr indicates poor prognosis for breast cancer and promotes cell proliferation and invasion, Eur. Rev. Med. Pharmacol. Sci., 22 (2018), 1358–1365. |
| [64] |
F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre, A. Jemal, Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA: Cancer J. Clinicians, 68 (2018), 394–424. https://doi.org/10.3322/caac.21492 doi: 10.3322/caac.21492
|
| [65] | Z. W. Wang, Y. Y. Jin, H. T. Ren, X. L. Ma, B. F. Wang, Y. L. Wang, Downregulation of the long non-coding RNA TUSC7 promotes NSCLC cell proliferation and correlates with poor prognosis, Am. J. Transl. Res., 8 (2016), 680–687. |
| [66] | H. P. Deng, L. Chen, T. Fan, B. Zhang, Y. Xu, Q. Geng, Long non-coding rna hottip promotes tumor growth and inhibits cell apoptosis in lung cancer, Cell. Mol. Biol., 61 (2015), 34–40. |
| [67] |
H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal, et al., Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J. Clinicians, 71 (2021), 209–249. https://doi.org/10.3322/caac.21660 doi: 10.3322/caac.21660
|
| [68] |
J. Q. Wang, L. P. Su, X. H. Chen, P. Li, Q. Cai, B. Q. Yu, et al., MALAT1 promotes cell proliferation in gastric cancer by recruiting SF2/ASF, Biomed. Pharmacother., 68 (2014), 557–564. https://doi.org/10.1016/j.biopha.2014.04.007 doi: 10.1016/j.biopha.2014.04.007
|
| [69] |
L. Ma, Y. J. Zhou, X. J. Luo, H. Gao, X. B. Deng, Y. J. Jiang, Long non-coding RNA XIST promotes cell growth and invasion through regulating miR-497/MACC1 axis in gastric cancer, Oncotarget, 8 (2017), 4125–4135. https://doi.org/10.18632/oncotarget.13670 doi: 10.18632/oncotarget.13670
|
| [70] |
H. T. Fu, F. Huang, X. Liu, Y. Qiu, W. Zhang, Mvgcn: data integration through multi-view graph convolutional network for predicting links in biomedical bipartite networks, Bioinformatics, 38 (2022), 426–434. https://doi.org/10.1093/bioinformatics/btab651 doi: 10.1093/bioinformatics/btab651
|









Figures(10) / Tables(6)

Li Peng, Yujie Yang, Cheng Yang, Zejun Li, Ngai Cheong. HRGCNLDA: Forecasting of lncRNA-disease association based on hierarchical refinement graph convolutional neural network[J]. Mathematical Biosciences and Engineering, 2024, 21(4): 4814-4834. doi: 10.3934/mbe.2024212







 DownLoad:
DownLoad: