Citation: Gaozhong Sun, Tongwei Zhao. Lung adenocarcinoma pathology stages related gene identification[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 737-746. doi: 10.3934/mbe.2020038

| [1] | D. S. Ettinger, W. Akerley, H. Borghaei, et al., Non-small cell lung cancer, version 2, 2013, J. Natl. Compr. Canc. Netw., 11 (2013), 645-653, quiz 653. |
| [2] | R. Siegel, D. Naishadham and A. Jemal, Cancer statistics, 2013, CA Cancer J. Clin., 63 (2013), 11-30. |
| [3] | D. E. Williams, P. C. Pairolero, C. S. Davis, et al., Survival of patients surgically treated for stage I lung cancer, J. Thorac. Cardiovasc. Surg., 82 (1981), 70-76. |
| [4] | G. A. Woodard, K. D. Jones and D. M. Jablons, Lung Cancer Staging and Prognosis, Cancer Treat. Res., 170 (2016), 47-75. |
| [5] | K. H. Yu, C. Zhang, G. J. Berry, et al., Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features, Nat. Commun., 7 (2016), 12474. |
| [6] | F. C. Detterbeck, D. J. Boffa and L. T. Tanoue, The new lung cancer staging system, Chest, 136 (2009), 260-271. |
| [7] | R. Diaz-Uriarte and S. Alvarez de Andres, Gene selection and classification of microarray data using random forest, BMC Bioinform., 7 (2006), 3. |
| [8] | V. A. Huynh-Thu, A. Irrthum, L. Wehenkel, et al., Inferring regulatory networks from expression data using tree-based methods, PLoS One, 5 (2010). |
| [9] | L. Breiman, Random Forests, Mach. Learn., 45 (2001). |
| [10] | S. Janitza, E. Celik and A. L. Boulesteix, A computationally fast variable importance test for random forests for high-dimensional data, Adv. Data Anal. Classific., (2016). |
| [11] | F. Degenhardt, S. Seifert and S. Szymczak, Evaluation of variable selection methods for random forests and omics data sets, Brief Bioinform., (2017). |
| [12] | A. Altmann, L. Tolosi, O. Sander, et al., Permutation importance: a corrected feature importance measure. Bioinformatics, 26 (2010), 1340-1347. |
| [13] | L. Wang, L. Yang, Z. Peng, et al., cisPath: an R/Bioconductor package for cloud users for visualization and management of functional protein interaction networks, BMC Syst. Biol., 9 (2015), S1. |
| [14] | A. Noto, S. Raffa, C. De Vitis, et al., Stearoyl-CoA desaturase-1 is a key factor for lung cancer-initiating cells, Cell Death Dis., 4 (2013), e947. |
| [15] | M. J. Cowley, K. S. Pinese, N. Kassahn, et al., PINA v2.0: Mining interactome modules, Nucleic Acids Res., 1 (2012), 40. |
| [16] | S. Razick, I. M. Magklaras and I. M. Donaldson, iRefIndex: A consolidated protein interaction database with provenance, BMC Bioinform., 9 (2008), 405. |
| [17] | A. Franceschini, S. Szklarczyk, M. Frankild, et al., STRING v9.1: Protein-protein interaction networks, with increased coverage and integration, Nucleic Acids Res., 11 (2013), 41. |
| [18] | C. Lemetre, Q. Zhang and Z. D. Zhang, SubNet: A Java application for subnetwork extraction, Bioinformatics, 29 (2013), 2509-2511. |
| [19] | P. Shannon, O. Markiel, N. S. Ozier, et al., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res., 13 (2003), 2498-2504. |
| [20] | Y. J. Kwon, S. J. Lee, J. S. Koh, et al., Genome-wide analysis of DNA methylation and the gene expression change in lung cancer, J. Thora. Onco., 7 (2012), 20-33. |
| [21] | J. Pan, G. Song, D. Chen, et al., Identification of serological biomarkers for early diagnosis of lung cancer using a protein array-based approach, Mol. Cell. Proteom., (2017). |
| [22] | E. E. Holmes, M. Jung, S. Meller, et al., Performance evaluation of kits for bisulfite-conversion of DNA from tissues, cell lines, FFPE tissues, aspirates, lavages, effusions, plasma, serum, and urine, PLoS One, 9 (2014), e93933. |
| [23] | V. Sailer, E. E. Holmes, H. Gevensleben, et al., PITX3 DNA methylation is an independent predictor of overall survival in patients with head and neck squamous cell carcinoma, Clin. Epigenet., 9 (2017), 12. |
| [24] | V. Sailer, E. E. Holmes, H. Gevensleben, et al., PITX2 and PANCR DNA methylation predicts overall survival in patients with head and neck squamous cell carcinoma. Oncotarget, 7 (2016), 75827-75838. |
| [25] | T. Fukagawa, A. Mikami, V. Nishihashi, et al., CENP-H, a constitutive centromere component, is required for centromere targeting of CENP-C in vertebrate cells, EMBO J., 20 (2001), 4603-4617. |
| [26] | N. Sugata, W. C. Li, T. J. Earnshaw, et al., Human CENP-H multimers colocalize with CENP-A and CENP-C at active centromere--kinetochore complexes, Hum. Mol. Genet., 9 (2000), 2919-2926. |
| [27] | W. T. Liao, X. Wang, L. H. Xu, et al., Centromere protein H is a novel prognostic marker for human nonsmall cell lung cancer progression and overall patient survival, Cancer, 115 (2009), 1507-1517. |
| [28] | H. R. Kim, G. O. Lee, K. H. Choi, et al., SRSF5: A novel marker for small-cell lung cancer and pleural metastatic cancer, Lung Cancer, 99 (2016), 57-65. |
| [29] | A. J. Lakhter, J. Hamilton, R. L. Konger, et al., Glucose-independent acetate metabolism promotes melanoma cell survival and tumor growth, J. Biol. Chem., 291 (2016), 21869-21879. |
| [30] | L. K. Boroughs and R. J. DeBerardinis, Metabolic pathways promoting cancer cell survival and growth, Nat. Cell. Biol., 17 (2015), 351-359. |
| [31] |
P. A. Watkins, D. Maiguel, Z. Jia, et al., Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome, J. Lipid. Res., 48 (2007), 2736-2750. doi: 10.1194/jlr.M700378-JLR200

|
| [32] | E. M. Kerr and C. P. Martins, Metabolic rewiring in mutant Kras lung cancer, FEBS J., 285 (2018), 28-41. |
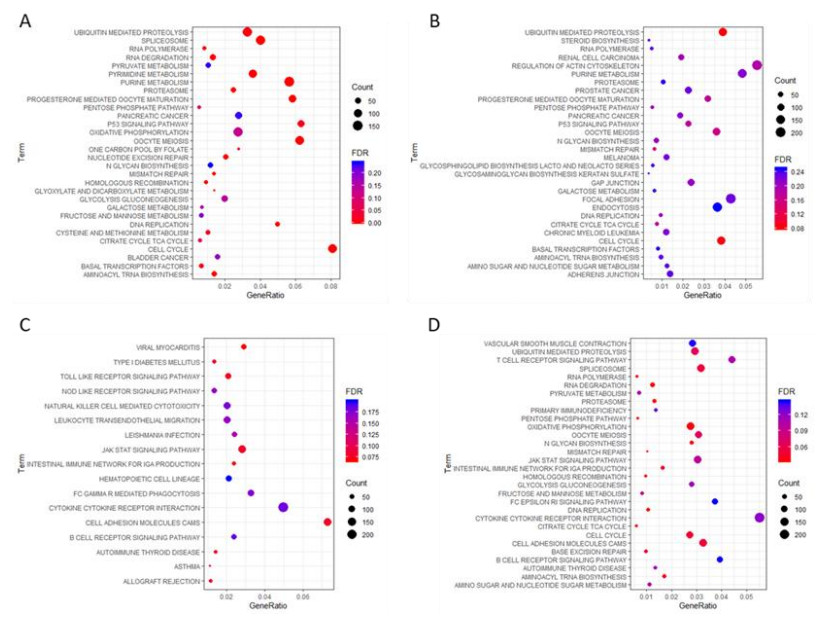
Figures(5) / Tables(2)

Gaozhong Sun, Tongwei Zhao. Lung adenocarcinoma pathology stages related gene identification[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 737-746. doi: 10.3934/mbe.2020038







 DownLoad:
DownLoad: