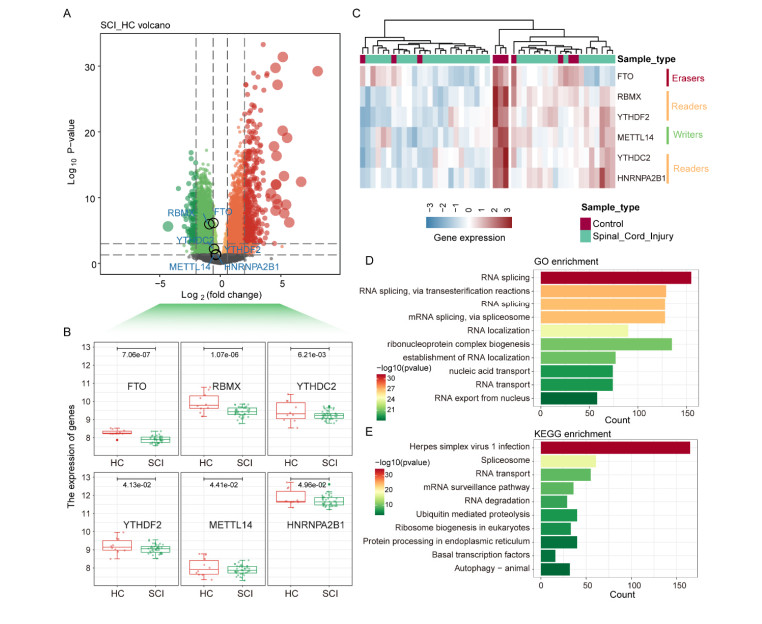
The diagnosis of the severity of spinal cord injury (SCI) and the revelation of potential therapeutic targets are crucial for urgent clinical care and improved patient outcomes. Here, we analyzed the overall gene expression data in peripheral blood leukocytes during the acute injury phase collected from Gene Expression Omnibus (GEO) and identified six m6A regulators specifically expressed in SCI compared to normal samples. LncRNA-mRNA network analysis identified AKT2/3 and PIK3R1 related to m6A methylation as potential therapeutic targets for SCI and constructed a classifier to identify patients of SCI to assist clinical diagnosis. Moreover, FTO (eraser) and RBMX (reader) were found to be significantly down-regulated in SCI and the functional gene co-expressed with them was found to be involved in the signal transduction of multiple pathways related to nerve injury. Through the construction of the drug-target gene network, eight key genes were identified as drug targets and it was emphasized that fostamatinib can be used as a potential drug for the treatment of SCI. Taken together, our study characterized the pathogenesis and identified a potential therapeutic target of SCI providing theoretical support for the development of precision medicine.
Citation: Shanzheng Wang, Xinhui Xie, Chao Li, Jun Jia, Changhong Chen. Integrative network analysis of N6 methylation-related genes reveal potential therapeutic targets for spinal cord injury[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 8174-8187. doi: 10.3934/mbe.2021405
The diagnosis of the severity of spinal cord injury (SCI) and the revelation of potential therapeutic targets are crucial for urgent clinical care and improved patient outcomes. Here, we analyzed the overall gene expression data in peripheral blood leukocytes during the acute injury phase collected from Gene Expression Omnibus (GEO) and identified six m6A regulators specifically expressed in SCI compared to normal samples. LncRNA-mRNA network analysis identified AKT2/3 and PIK3R1 related to m6A methylation as potential therapeutic targets for SCI and constructed a classifier to identify patients of SCI to assist clinical diagnosis. Moreover, FTO (eraser) and RBMX (reader) were found to be significantly down-regulated in SCI and the functional gene co-expressed with them was found to be involved in the signal transduction of multiple pathways related to nerve injury. Through the construction of the drug-target gene network, eight key genes were identified as drug targets and it was emphasized that fostamatinib can be used as a potential drug for the treatment of SCI. Taken together, our study characterized the pathogenesis and identified a potential therapeutic target of SCI providing theoretical support for the development of precision medicine.

| [1] | J. W. McDonald, C. Sadowsky, Spinal-cord injury, Lancet, 359 (2002), 417-425. |
| [2] |
J. C. Furlan, V. Noonan, A. Singh, M. G. Fehlings, Assessment of impairment in patients with acute traumatic spinal cord injury: a systematic review of the literature, J. Neurotrauma, 28 (2011), 1445-1477. doi: 10.1089/neu.2009.1152

|
| [3] | C. S. Ahuja, J. R. Wilson, S. Nori, M. R. Kotter, C. Druschel, A. Curt, et al., Traumatic spinal cord injury, Nat. Rev. Dis. Primers, 3 (2017), 1-21. |
| [4] |
S. B. Lim, W. D. Lee, J. Vasudevan, W. T. Lim, C. T. Lim, Liquid biopsy: one cell at a time, NPJ Precis. Oncol., 3 (2019), 1-9. doi: 10.1038/s41698-018-0074-x
|
| [5] | S. Hocine, R. H. Singer, D. Grunwald, RNA processing and export, CSH. Perspect. Biol., 2 (2010), a000752. |
| [6] | M. Francois, P. Donovan, F. Fontaine, Modulating transcription factor activity: Interfering with protein-protein interaction networks, Semin. Cell Dev. Biol., 99 (2020) 12-19. |
| [7] | Y. Zhang, P. Han, Q. Guo, Y. Hao, Y. Qi, M. Xin, et al., Oncogenic landscape of somatic mutations perturbing pan-cancer lncRNA-ceRNA regulation, Front. Cell Dev. Biol., 9 (2021) 658346. |
| [8] |
N. Liu, Q. Dai, G. Zheng, C. He, M. Parisien, T. Pan, N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions, Nature, 518 (2015), 560-564. doi: 10.1038/nature14234
|
| [9] |
P. K. Yadav, R. Rajasekharan, The m(6)A methyltransferase Ime4 epitranscriptionally regulates triacylglycerol metabolism and vacuolar morphology in haploid yeast cells, J. Biol. Chem., 292 (2017), 13727-13744. doi: 10.1074/jbc.M117.783761
|
| [10] |
D. P. Patil, B. F. Pickering, S. R. Jaffrey, Reading m(6)A in the transcriptome: m(6)A-binding proteins, Trends Cell Biol., 28 (2018), 113-127. doi: 10.1016/j.tcb.2017.10.001
|
| [11] |
Y. Fu, D. Dominissini, G. Rechavi, C. He, Gene expression regulation mediated through reversible m(6)A RNA methylation, Nat. Rev. Genet., 15 (2014), 293-306. doi: 10.1038/nrg3724
|
| [12] | T. Barrett, S. E. Wilhite, P. Ledoux, C. Evangelista, I. F. Kim, M. Tomashevsky, et al., NCBI GEO: archive for functional genomics data sets--update, Nucleic Acids Res., 41 (2012), D991-D995. |
| [13] |
Y. Li, J. Xiao, J. Bai, Y. Tian, Y. Qu, X. Chen, et al., Molecular characterization and clinical relevance of m(6)A regulators across 33 cancer types, Mol. Cancer, 18 (2019), 1-6. doi: 10.1186/s12943-018-0930-x
|
| [14] | H. Hu, Y. R. Miao, L. H. Jia, Q. Y. Yu, Q. Zhang, A. Y. Guo, AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors, Nucleic Acids Res., 47 (2019), D33-D38. |
| [15] | H. Han, J. W. Cho, S. Lee, A. Yun, H. Kim, D. Bae, et al., TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions, Nucleic Acids Res., 46 (2018), D380-D386. |
| [16] | F. Vafaee, J. R. Krycer, X. Ma, T. Burykin, D. E. James, Z. Kuncic, ORTI: An open-access repository of transcriptional interactions for interrogating mammalian gene expression data, Plos One, 11 (2016), e0164535. |
| [17] | J. H. Li, S. Liu, H. Zhou, L. H. Qu, J. H. Yang, starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data, Nucleic Acids Res., 42 (2014), D92- D97. |
| [18] | A. Frankish, M. Diekhans, A. M. Ferreira, R. Johnson, I. Jungreis, J. Loveland, et al., Gencode reference annotation for the human and mouse genomes, Nucleic Acids Res., 47 (2019), D766-D773. |
| [19] |
A. Liberzon, A. Subramanian, R. Pinchback, H. Thorvaldsdottir, P. Tamayo, J.P. Mesirov, Molecular signatures database (MSigDB) 3.0, Bioinformatics, 27 (2011), 1739-1740. doi: 10.1093/bioinformatics/btr260
|
| [20] | D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, et al., DrugBank 5.0: a major update to the DrugBank database for 2018, Nucleic Acids Res., 46 (2018), D1074-D1082. |
| [21] |
M. I. Love, W. Huber, S. Anders, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol., 15 (2014), 1-21. doi: 10.1186/gb-2014-15-1-r1
|
| [22] |
F. Degenhardt, S. Seifert, S. Szymczak, Evaluation of variable selection methods for random forests and omics data sets, Brief. Bioinform., 20 (2019), 492-503. doi: 10.1093/bib/bbx124
|
| [23] |
P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, et al., Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Res., 13 (2003), 2498-2504. doi: 10.1101/gr.1239303
|
| [24] | S. Hanzelmann, R. Castelo, J. Guinney, GSVA: gene set variation analysis for microarray and RNA-seq data, BMC Bioinformatics, 14 (2013), 1-15. |
| [25] |
M. D. Wilkerson, D.N. Hayes, ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking, Bioinformatics, 26 (2010), 1572-1573. doi: 10.1093/bioinformatics/btq170
|
| [26] |
I. Cervellini, J. Galino, N. Zhu, S. Allen, C. Birchmeier, D. L. Bennett, Sustained MAPK/ERK activation in adult Schwann cells impairs nerve repair, J. Neurosci., 38 (2018), 679-690. doi: 10.1523/JNEUROSCI.2255-17.2017
|
| [27] | H. Mohammed, L. Rimondini, V. Rocchetti, Molecular basis of trigeminal nerve disorders and healing, Eur Rev Med Pharmacol Sci, 22 (2018), 5755-5764. |
| [28] |
D. Gao, T. Tang, J. Zhu, Y. Tang, H. Sun, S. Li, CXCL12 has therapeutic value in facial nerve injury and promotes Schwann cells autophagy and migration via PI3K-AKT-mTOR signal pathway, Int. J. Biol. Macromol., 124 (2019), 460-468. doi: 10.1016/j.ijbiomac.2018.10.212
|
| [29] |
A. K. Patel, K. K. Park, A. S. Hackam, Wnt signaling promotes axonal regeneration following optic nerve injury in the mouse, Neuroscience, 343 (2017), 372-383. doi: 10.1016/j.neuroscience.2016.12.020
|
| [30] |
C. Shen, B. Xuan, T. Yan, Y. Ma, P. Xu, X. Tian, et al., m(6)A-dependent glycolysis enhances colorectal cancer progression, Mol. Cancer, 19 (2020), 1-19. doi: 10.1186/s12943-019-1085-0
|
| [31] |
G. Yu, L. G. Wang, Y. Han, Q. Y. He, ClusterProfiler: an R package for comparing biological themes among gene clusters, OMICS., 16 (2012), 284-287. doi: 10.1089/omi.2011.0118
|
| [32] |
X. Qian, J. Zhao, P. Y. Yeung, Q. C. Zhang, C. K. Kwok, Revealing lncRNA structures and interactions by sequencing-based approaches, Trends Biochem. Sci., 44 (2019), 33-52. doi: 10.1016/j.tibs.2018.09.012
|
| [33] |
L. Li, L. Wang, H. Li, X. Han, S. Chen, B. Yang, et al., Characterization of lncRNA expression profile and identification of novel lncRNA biomarkers to diagnose coronary artery disease, Atherosclerosis, 275 (2018), 359-367. doi: 10.1016/j.atherosclerosis.2018.06.866
|
| [34] |
Y. Xia, L. Zhen, H. Li, S. Wang, S. Chen, C. Wang, et al., MIRLET7BHG promotes hepatocellular carcinoma progression by activating hepatic stellate cells through exosomal SMO to trigger Hedgehog pathway, Cell Death Dis., 12 (2021), 1-17. doi: 10.1038/s41419-020-03229-8
|
| [35] |
N. Moreau, Y. Boucher, Hedging against neuropathic pain: role of hedgehog signaling in pathological nerve healing, Int. J. Mol. Sci., 21 (2020), 9115. doi: 10.3390/ijms21239115
|
| [36] |
N. Zhang, X. Zeng, C. Sun, H. Guo, T. Wang, L. Wei, et al., LncRNA linc00963 promotes tumorigenesis and radioresistance in breast cancer by sponging miR-324-3p and inducing ack1 expression, Mol. Ther. Nucl. Acids, 18 (2019), 871-881. doi: 10.1016/j.omtn.2019.09.033
|
| [37] | B. Q. Qiu, X. H. Lin, X. D. Ye, W. Huang, X. Pei, D. Xiong, et al., Long non-coding RNA PSMA3-AS1 promotes malignant phenotypes of esophageal cancer by modulating the miR-101/EZH2 axis as a ceRNA, Aging (Albany NY), 12 (2020), 1843-1856. |
| [38] |
K. A. Papavassiliou, A. G. Papavassiliou, Transcription factor drug targets, J. Cell. Biochem., 117 (2016), 2693-2696. doi: 10.1002/jcb.25605
|
| [39] |
M. Hecker, A. H. Wagner, Transcription factor decoy technology: A therapeutic update, Biochem. Pharmacol., 144 (2017), 29-34. doi: 10.1016/j.bcp.2017.06.122
|
| [40] |
Y. L. Liu, L. J. Zhou, N. W. Hu, J. T. Xu, C. Y. Wu, T. Zhang, et al., Tumor necrosis factor-alpha induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK, Neuropharmacology, 52 (2007), 708-15. doi: 10.1016/j.neuropharm.2006.09.011
|
| [41] |
T. Csepany, A. Lin, C. J. Baldick, K. Beemon, Sequence specificity of mRNA N6-adenosine methyltransferase, J. Biol. Chem., 265 (1990), 20117-20122. doi: 10.1016/S0021-9258(17)30477-5
|
| [42] |
M. Chen, C. M. Wong, The emerging roles of N6-methyladenosine (m6A) deregulation in liver carcinogenesis, Mol. Cancer, 19 (2020), 1-12. doi: 10.1186/s12943-019-1085-0
|
| [43] |
Z. Li, H. Weng, R. Su, X. Weng, Z. Zuo, C. Li, et al., FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase, Cancer Cell, 31 (2017), 127-141. doi: 10.1016/j.ccell.2016.11.017
|
| [44] | E. S. Fu, Y. P. Zhang, J. Sagen, K. A. Candiotti, P. D. Morton, D. J. Liebl, et al., Transgenic inhibition of glial NF-kappa B reduces pain behavior and inflammation after peripheral nerve injury, Pain, 148 (2010), 509-518. |
 mbe-18-06-405 supplyment .docx mbe-18-06-405 supplyment .docx |
 |




Figures(5)

Shanzheng Wang, Xinhui Xie, Chao Li, Jun Jia, Changhong Chen. Integrative network analysis of N6 methylation-related genes reveal potential therapeutic targets for spinal cord injury[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 8174-8187. doi: 10.3934/mbe.2021405







 DownLoad:
DownLoad: