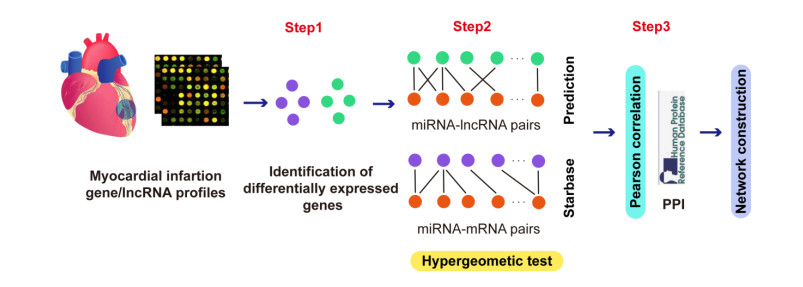
Myocardial infarction (MI) is a type of coronary heart disease, which refers to the ischemic necrosis of the heart muscle. A large number of studies have discussed the mechanism of MI from the perspective of competing endogenous RNA (ceRNA) network. However, the mechanisms underlying the function of lncRNAs in MI have still not been explained in an explicit manner. Therefore, we constructed a scale-free lncRNA-associated ceRNA network to identify some crucial lncRNAs in MI. Results showed that the given disease genes for MI were involved in the network, the degrees of which were significantly larger than the other nodes of the network. For measuring the network centrality, we then constructed a hub subnetwork. The miRNAs and mRNAs in the hub subnetwork have been validated to function in MI-related biological function. In addition, we identified 2 MI-related functional modules from the lncRNA-associated ceRNA network, which suggested that lncRNA exerted function in local network. Enrichment analysis showed that these functional modules corresponded to some similar and different pathways related to cardiovascular disease. More importantly, 3 MI-related crucial lncRNAs, CTD-3092A11.2, RP5-821D11.7 and CTC-523E23.1 were detected as potential biomarkers, which may be involved in MI-related biological progresses. Our study identified 20 functional lncRNAs based on ceRNA network analysis, which may provide novel diagnosis and therapeutic targets for MI from the ceRNA network perspective.
Citation: Beibei Zhu, Yue Mao, Mei Li. Identification of functional lncRNAs through constructing a lncRNA-associated ceRNA network in myocardial infarction[J]. Mathematical Biosciences and Engineering, 2021, 18(4): 4293-4310. doi: 10.3934/mbe.2021215
Myocardial infarction (MI) is a type of coronary heart disease, which refers to the ischemic necrosis of the heart muscle. A large number of studies have discussed the mechanism of MI from the perspective of competing endogenous RNA (ceRNA) network. However, the mechanisms underlying the function of lncRNAs in MI have still not been explained in an explicit manner. Therefore, we constructed a scale-free lncRNA-associated ceRNA network to identify some crucial lncRNAs in MI. Results showed that the given disease genes for MI were involved in the network, the degrees of which were significantly larger than the other nodes of the network. For measuring the network centrality, we then constructed a hub subnetwork. The miRNAs and mRNAs in the hub subnetwork have been validated to function in MI-related biological function. In addition, we identified 2 MI-related functional modules from the lncRNA-associated ceRNA network, which suggested that lncRNA exerted function in local network. Enrichment analysis showed that these functional modules corresponded to some similar and different pathways related to cardiovascular disease. More importantly, 3 MI-related crucial lncRNAs, CTD-3092A11.2, RP5-821D11.7 and CTC-523E23.1 were detected as potential biomarkers, which may be involved in MI-related biological progresses. Our study identified 20 functional lncRNAs based on ceRNA network analysis, which may provide novel diagnosis and therapeutic targets for MI from the ceRNA network perspective.

| [1] | T. S. Hartikainen, N. A. Sorensen, P. M. Haller, A. Gossling, J. Lehmacher, T. Zeller, et al., Clinical application of the 4th universal definition of myocardial infarction, Europ. Heart J., 2020. |
| [2] |
D. A. Morrow, The fourth universal definition of myocardial infarction and the emerging importance of myocardial injury, Circulation, 141 (2020), 172. doi: 10.1161/CIRCULATIONAHA.119.044125

|
| [3] | A. Singh, A. Gupta, E. M. DeFilippis, A. Qamar, D. W Biery, Z. Almarzooq, et al., Cardiovascular mortality after type 1 and type 2 myocardial infarction in young adults, J. Am. Coll. Cardiol., 75 (2020), 1003-1013. |
| [4] |
C. Y. Liu, Y. H. Zhang, R. B. Li, L. Y. Zhou, T. An, R. C. Zhang, et al., LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription, Nat. Commun., 9 (2018), 29. doi: 10.1038/s41467-017-02280-y
|
| [5] |
K. Wang, C. Y. Liu, L. Y. Zhou, J. X. Wang, M. Wang, B. Zhao, et al., APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p, Nat. Commun., 6 (2015), 6779. doi: 10.1038/ncomms7779
|
| [6] |
Y. Zhang, L. Jiao, L. Sun, Y. Li, Y. Gao, C. Xu, et al., LncRNA ZFAS1 as a SERCA2a inhibitor to cause intracellular Ca(2+) overload and contractile dysfunction in a mouse model of myocardial infarction, Circul. Res., 122 (2018), 1354-1368. doi: 10.1161/CIRCRESAHA.117.312117
|
| [7] | L. Salmena, L. Poliseno, Y. Tay, L. Kats, P. P. Pandolfi, A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell, 146 (2011), 353-358. |
| [8] | G. Zhang, H. Sun, Y. Zhang, H. Zhao, W. Fan, J. Li, et al., Characterization of dysregulated lncRNA-mRNA network based on ceRNA hypothesis to reveal the occurrence and recurrence of myocardial infarction, Cell Death Discovery, 4 (2018), 35. |
| [9] |
Q. Mao, X. L. Liang, C. L. Zhang, Y. H. Pang, Y. X. Lu, LncRNA KLF3-AS1 in human mesenchymal stem cell-derived exosomes ameliorates pyroptosis of cardiomyocytes and myocardial infarction through miR-138-5p/Sirt1 axis, Stem. Cell Res. Ther., 10 (2019), 393. doi: 10.1186/s13287-019-1522-4
|
| [10] |
C. Song, J. Zhang, Y. Liu, H. Pan, H. P. Qi, Y. G. Cao, et al, Construction and analysis of cardiac hypertrophy-associated lncRNA-mRNA network based on competitive endogenous RNA reveal functional lncRNAs in cardiac hypertrophy, Oncotargets, 7 (2016), 10827-10840. doi: 10.18632/oncotarget.7312
|
| [11] |
D. Vella, S. Marini, F. Vitali, D. D. Silvestre, G. Mauri, R. Bellazzi, MTGO: PPI network analysis via topological and functional module identification, Sci. Rep., 8 (2018), 5499. doi: 10.1038/s41598-018-29025-1
|
| [12] | G. D. Bader, C. W. Hogue, An automated method for finding molecular complexes in large protein interaction networks, BMC Bioinf., 4 (2003). |
| [13] |
B. Adamcsek, G. Palla, I. J. Farkas, I. Derenyi, T. Vicsek, CFinder: locating cliques and overlapping modules in biological networks, Bioinformatics, 22 (2006), 1021-1023. doi: 10.1093/bioinformatics/btl039
|
| [14] |
J. C. Smoot, K. D. Barbian, J. J. V. Gompel, L. M. Smoot, M. S. Chaussee, G.. Sylva, et al., Genome sequence and comparative microarray analysis of serotype M18 group A Streptococcus strains associated with acute rheumatic fever outbreaks, Proc. Nat. Acad. Sci. U. S. A., 99 (2002), 4668-4673. doi: 10.1073/pnas.062526099
|
| [15] | Y. Li, X. N. He, C. Li, L. Gong, M.Liu, Identification of candidate genes and microRNAs for acute myocardial infarction by weighted gene coexpression network analysis, Biomed. Res. Int. 2019 (2019), 5742608. |
| [16] |
L. Wang, W. Yuan, J. Huang, Identification of myocardial infarction-associated genes using integrative microRNA-gene expression network analysis, DNA Cell. Biol., 40 (2021), 348-358. doi: 10.1089/dna.2020.6222
|
| [17] | J. H. Li, S. Liu, H. Zhou, L. H. Qu, J. H. Yang, StarBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data, Nucleic Acids Res., 42 (2014), D92-D97. |
| [18] | J. Pinero, J. M. Ramirez-Anguita, J.Sauch-Pitarch, F. Ronzano, E. Centeno, F. Sanz, et al., The DisGeNET knowledge platform for disease genomics: 2019 update, Nucleic Acids Res., 48 (2020), D845-D855. |
| [19] |
S. Zhang, X. Liu, S.Goldstein, Y. Li, J. Ge, B. He, et al, Role of the JAK/STAT signaling pathway in the pathogenesis of acute myocardial infarction in rats and its effect on NF-kappaB expression, Mol. Med. Rep., 7 (2013), 93-98. doi: 10.3892/mmr.2012.1159
|
| [20] | J. Zhu, H. Gu, X. Lv, C. Yuan, P. Ni, F. Liu, LINC-PINT activates the mitogen-activated protein kinase pathway to promote acute myocardial infarction by regulating miR-208a-3p, Circ. J.: Off. J. Jpn. Circ. Soc., 82 (2018), 2783-2792. |
| [21] | J. H. Li, J. Dai, B. Han, G. H. Wu, C. H. Wang, MiR-34a regulates cell apoptosis after myocardial infarction in rats through the Wnt/beta-catenin signaling pathway, Europ. Rev. Med. Pharmacol. Sci., 23 (2019), 2555-2562. |
| [22] | P. M. Voorhoeve, C. le Sage, M. Schrier, A. J. Gillis, H. Stoop, R. Nagel, et al., A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors, Cell, 126 (2006), 1169-1181. |
| [23] |
F. Wei, C. Cao, X. Xu, J. Wang, Diverse functions of miR-373 in cancer, J. Transl. Med., 13 (2015), 162. doi: 10.1186/s12967-015-0523-z
|
| [24] |
J. Liu, M. Jiang, S. Deng, J. Lu, H. Huang, Y. Zhang, et al., MiR-93-5p-containing exosomes treatment attenuates acute myocardial infarction-induced myocardial damage, Mol. Ther. Nucleic Acids, 11 (2018), 103-115. doi: 10.1016/j.omtn.2018.01.010
|
| [25] | D. Ke, Q. Wang, S. Ke, L. Zou, Q. Wang, Long-non coding RNA SNHG16 supports colon cancer cell growth by modulating miR-302a-3p/AKT axis, Pathol. Oncol. Res. : POR, 2019. |
| [26] | L. Tai, C. J. Huang, K. B. Choo, S. K. Cheong, T. Kamarul, Oxidative stress down-regulates MiR-20b-5p, MiR-106a-5p and E2F1 expression to suppress the G1/S transition of the cell cycle in multipotent stromal cells, Int. J. Med. Sci., 17 (2020), 457-470. |
| [27] |
Y. Yu, J. Zhang, Y. Jin, Y. Yang, J. Shi, F. Chen, et al., MiR-20a-5p suppresses tumor proliferation by targeting autophagy-related gene 7 in neuroblastoma, Cancer Cell Int., 18 (2018), 5. doi: 10.1186/s12935-017-0499-2
|
| [28] |
Y. Hu, G. Jin, B. Li, Y. Chen, L. Zhong, G. Chen, et al., Suppression of miRNA let-7i-5p promotes cardiomyocyte proliferation and repairs heart function post injury by targetting CCND2 and E2F2, Clin. Sci., 133 (2019), 425-441. doi: 10.1042/CS20181002
|
| [29] |
M. Dziemidowicz, T. A. Bonda, S. Litvinovich, A. Taranta, M. M. Winnicka, K. A. Kaminski, The role of interleukin-6 in intracellular signal transduction after chronic beta-adrenergic stimulation in mouse myocardium, Arch. Med. Sci.: AMS, 15 (2019), 1565-1575. doi: 10.5114/aoms.2019.89452
|
| [30] |
B. C. Bernardo, X. M. Gao, C. E. Winbanks, E. J. Boey, Y. K. Tham, H. Kiriazis, et al., Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function, Proc. Nat. Acad. Sci. U. S. A., 109 (2012), 17615-176120. doi: 10.1073/pnas.1206432109
|
| [31] | Y. Wang, Y. Huang, M. Zhang, X. Zhang, X. Tang, Y. Kang, Bioinformatic analysis of the possible regulative network of miR-30a/e in cardiomyocytes 2 days post myocardial infarction, Acta Cardiol. Sin., 34 (2018), 175-188. |
| [32] |
L. S. Shen, X. F. Hu, T. Chen, G. L. Shen, D. Cheng, Integrated network analysis to explore the key mRNAs and lncRNAs in acute myocardial infarction, Math. Biosci. Eng., 16 (2019), 6426-6437. doi: 10.3934/mbe.2019321
|
| [33] |
K. Wu, Q. Zhao, Z. Li, N. Li, Q. Xiao, X. Li, et al., Bioinformatic screening for key miRNAs and genes associated with myocardial infarction, FEBS Open Bio., 8 (2018), 897-913. doi: 10.1002/2211-5463.12423
|
| [34] |
I. Keklikoglou, C. Koerner, C. Schmidt, J. D. Zhang, D. Heckmann, A. Shavinskaya, et al., MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting NF-kappaB and TGF-beta signaling pathways, Oncogene, 31 (2012), 4150-4163. doi: 10.1038/onc.2011.571
|
| [35] |
A. D. Zhou, L. T. Diao, H. Xu, Z. D. Xiao, J. H. Li, H. Zhou, et al., Beta-Catenin/LEF1 transactivates the microRNA-371-373 cluster that modulates the Wnt/beta-catenin-signaling pathway, Oncogene, 31 (2012), 2968-2978. doi: 10.1038/onc.2011.461
|
| [36] |
A. Mukherjee, A. M. D. Bisceglie, R. B. Ray, Hepatitis C virus-mediated enhancement of microRNA miR-373 impairs the JAK/STAT signaling pathway, J. Virol., 89 (2015), 3356-3365. doi: 10.1128/JVI.03085-14
|
| [37] |
I. Nikolayeva, O. G. Pla, B. Schwikowski, Network module identification-A widespread theoretical bias and best practices, Methods, 132 (2018), 19-25. doi: 10.1016/j.ymeth.2017.08.008
|
| [38] |
C. A. Makarewich, H. Zhang, J. Davis, R. N. Correll, D. M. Trappanese, N. E. Hoffman, et al., Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction, Circ. Res., 115 (2014), 567-580. doi: 10.1161/CIRCRESAHA.115.303831
|
| [39] |
K. V. der Borght, C. L. Scott, V. Nindl, A. Bouche, L. Martens, D. Sichien, et al., Myocardial infarction primes autoreactive T cells through activation of dendritic cells, Cell Rep., 18 (2017), 3005-3017. doi: 10.1016/j.celrep.2017.02.079
|
| [40] |
P. Ortiz-Sanchez, M. Villalba-Orero, M. M. Lopez-Olaneta, J. Larrasa-Alonso, F. Sanchez-Cabo, C. Marti-Gomez, et al., Loss of SRSF3 in cardiomyocytes leads to decapping of contraction-related mRNAs and severe systolic dysfunction, Circ. Res., 125 (2019), 170-183. doi: 10.1161/CIRCRESAHA.118.314515
|
| [41] |
J. W. Waks, A. E. Buxton, Risk stratification for sudden cardiac death after myocardial infarction, Ann. Rev. Med., 69 (2018), 147-164. doi: 10.1146/annurev-med-041316-090046
|
| [42] |
J. Peng, L. Zhang, C. Yuan, L. Zhou, S. Xu, Y. Lin, et al., Expression profile analysis of long noncoding RNA in ER-positive subtype breast cancer using microarray technique and bioinformatics, Cancer Manage. Res., 9 (2017), 891-901. doi: 10.2147/CMAR.S151120
|
| [43] |
B. F. Zhang, H. Jiang, J. Chen, Q. Hu, S. Yang, X. P. Liu, et al., LncRNA H19 ameliorates myocardial infarction-induced myocardial injury and maladaptive cardiac remodelling by regulating KDM3A, J. Cell. Mol. Med. 24 (2020), 1099-1115. doi: 10.1111/jcmm.14846
|
| [44] | H. Liang, X. Su, Q.Wu, H. Shan, L. Lv, T. Yu, et al., LncRNA 2810403D21Rik/Mirf promotes ischemic myocardial injury by regulating autophagy through targeting Mir26a, Autophagy, (2019), 1-15. |







Figures(8) / Tables(1)

Beibei Zhu, Yue Mao, Mei Li. Identification of functional lncRNAs through constructing a lncRNA-associated ceRNA network in myocardial infarction[J]. Mathematical Biosciences and Engineering, 2021, 18(4): 4293-4310. doi: 10.3934/mbe.2021215







 DownLoad:
DownLoad: