Citation: Samantha Erwin, Stanca M. Ciupe. Germinal center dynamics during acute and chronic infection[J]. Mathematical Biosciences and Engineering, 2017, 14(3): 655-671. doi: 10.3934/mbe.2017037

| [1] | [ C. Allen,T. Okada,J. Cyster, Germinal-center organization and cellular dynamcs, Immunity, 27 (2007): 190-202. |
| [2] | [ B. Asquith,C. Debacq,A. Florins,N. Gillet,T. Sanchez-Alcaraz,A. Mosley,L. Willems, Quantifying lymphocyte kinetics in vivo using carboxyfluorescein diacetate succinimidyl ester (CFSE), Proc Biol Sci, 273 (2006): 1165-1171. |
| [3] | [ D. Burton,J. Mascola, Antibody responses to envelope glycoproteins in HIV-1 infection, Nat Immunol, 16 (2015): 571-576. |
| [4] | [ R. Cubas,J. Mudd,A. Savoye,M. Perreau,J. van Grevenynghe, Inadequate T follicular cell help impairs B cell immunity during HIV infection, Nat Med, 19 (2013): 494-499. |
| [5] | [ R. De Boer,A. Perelson, Quantifying T lymphocyte turnover, J Theor Biol, 327 (2013): 45-87. |
| [6] | [ A. Hauser,M. Shlomchik,A. Haberman, In vivo imaging studies shed light on germinal-centre development, Nat Rev Immunol, 7 (2007): 499-504. |
| [7] | [ B. Haynes, New approaches to HIV vaccine development, Curr Opin Immunol, 35 (2015): 39-47. |
| [8] | [ P. Hodgkin,J. Lee,A. Lyons, B cell differentiation and isotype switching is related to division cycle number, J Exp Med, 184 (1996): 277-281. |
| [9] | [ K. Hollowood,J. Macartney, Cell kinetics of the germinal center reaction -a stathmokinetic study, Eur J Immunol, 22 (1992): 261-266. |
| [10] | [ T. Kepler,A. Perelson, Cyclic re-entry of germinal center B cells and the efficiency of affinity maturations, Immunol Today, 14 (1993): 412-415. |
| [11] | [ C. Kesmir,R. De Boer, A mathematical model on germinal center kinetics and termination, J Immunol, 163 (1999): 2463-2469. |
| [12] | [ C. Kesmir,R. de Boer, A spatial model of germinal center reactions: Cellular adhesion based sorting of B cells results in efficient affinity maturation, J Theor Biol, 222 (2003): 9-22. |
| [13] | [ F. Kroese,A. Wubbena,H. Seijen,P. Nieuwenhuis, Germinal centers develop oligoclonally, Eur J Immunol, 17 (1987): 1069-1072. |
| [14] | [ R. Kuppers,M. Zhao,M. Hansmann,K. Rajewsky, Tracing B cell development in human germinal centers by molecular analysis of single cells picked from histological sections, Embo J, 12 (1993): 4955-4967. |
| [15] | [ P. Kwong,J. Mascola, Human antibodies that neutralize HIV-1: Identification, structures, and B cell ontogenies, Immunity, 37 (2012): 412-425. |
| [16] | [ V. L,M. Diaz, Autoreactivity in HIV-1 broadly neutralizing antibodies: Implications for their function and induction by vaccination, Curr Opin HIV AIDS, 9 (2014): 224-234. |
| [17] | [ H. Lee,E. Hawkins,M. Zand,T. Mosmann,H. Wu, Interpreting CFSE obtained division histories of B cells in vitro with Smith-Martin and cyton type models, Bull Math Biol, 71 (2009): 1649-1670. |
| [18] | [ M. Lindqvist,J. van Lunzen,D. Soghoian,B. Kuhl,S. Ranasinghe, Expansion of HIV-specific T follicular helper cells in chronic HIV infection, J Clin Invest, 122 (2012): 3271-3280. |
| [19] | [ I. MacLennan, Germinal centers, Annu Rev Immunol., 12 (1994): 117-139. |
| [20] | [ M. Meyer-Hermann,E. Mohr,N. Pelletier,Y. Zhang,G. Victoria,K. Toellner, A theory of germinal center B cell selection, division, and exit, Cell Reports, 2 (2012): 162-174. |
| [21] | [ M. Meyer-Hermann,M. Figge,K. Toellner, Germinal centres seen through the mathematical eye: B-cell models on the catwalk, Trends in Immunol, 30 (2009): 157-164. |
| [22] | [ M. Meyer-Hermann,P. Maini, Cutting edge: Back to one-way germinal centers, J Immunol, 174 (2005): 2489-2493. |
| [23] | [ M. Meyer-Hermann,P. Maini,D. Iber, An analysis of B cell selection mechanisms in germinal centers, Math Med Biol, 23 (2006): 255-277. |
| [24] | [ H. Miao,X. Jin,A. Perelson,H. Wu, Evaluation of multitype mathematical models for CFSE-labeling experiment data, Bull Math Biol, 74 (2012): 300-326. |
| [25] | [ I. Mikell,D. Sather,S. Kalams,M. Altfeld,G. Alter,L. Stamatatos, Characteristics of the earliest cross-neutralizing antibody response to HIV-1, PLoS Pathog, 7 (2011): 1-15. |
| [26] | [ M. Moody,R. Zhang,E. Walter,C. Woods,G. Ginsburg, H3N2 influenza infection elicits more cross-reactive and less clonally expanded anti-hemagglutinin antibodies than influenza vaccination, PLoS One, 6 (2011): e25797, 14pp. |
| [27] | [ J. Moreira,J. Faro, Modelling two possible mechanisms for the regulation of the germinal center dynamics, J Immunol, 177 (2006): 3705-3710. |
| [28] | [ M. Oprea,A. Perelson, Somatic mutation leads to efficient affinity maturation when centrocytes recycle back to centroblasts, J Immunol, 158 (1997): 5155-5162. |
| [29] | [ M. Oprea,E. van Nimwegen,A. Perelson, Dynamics of one-pass germinal center models: Implications for affinity maturation, Bull Math Biol, 62 (2000): 121-153. |
| [30] | [ S. Pallikkuth,A. Parmigiani,S. Pahwa, The role of interleukin-21 in HIV infection, Cytokine growth factor rev, 23 (2012): 173-180. |
| [31] | [ M. Perreau,A.-L. Savoye,E. De Crignis,J. Corpataux,R. Cubas, Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production, J Exp Med, 210 (2013): 143-156. |
| [32] | [ J. Publicover,A. Gaggar,S. Nishimura,C. Van Horn,A. Goodsell, Age-dependent hepatic lymphoid organization directs successful immunity to hepatitis B, J Clin Invest, 123 (2013): 3728-3739. |
| [33] | [ J. Publicover,A. Goodsell,S. Nishimura,S. Vilarinho,Z. Wang, IL-21 is pivotal in determining age-dependent effectiveness of immune responses in a mouse model of human hepatitis B, J Clin Invest, 121 (2011): 1154-1162. |
| [34] | [ M. Radmacher,G. Kelsoe,T. Kepler, Predicted and inferred waiting times for key mutations in the germinal centre reaction: evidence for stochasticity in selection, Immunol and Cell Bio, 76 (1998): 373-381. |
| [35] | [ T. Schwickert,G. Victoria,D. Fooksman,A. Kamphorst,M. Mugnier, A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center, J Exp Med, 208 (2011): 1243-1252. |
| [36] | [ Z. Shulman,A. Gitlin,S. Targ,M. Jankovic,G. Pasqual, T follicular helper cell dynamics in germinal centers, Science, 341 (2013): 673-677. |
| [37] | [ G. Siskind,B. Benacerraf, Cell selection by antigen in the immune response, Adv. Immunol., 10 (1969): 1-50. |
| [38] | [ M. Stafford,L. Corey,Y. Cao,E. Daar,D. Ho,A. Perelson, Modeling plasma virus concentration during primary HIV infection, J theor Biol, 203 (2000): 285-301. |
| [39] | [ L. Stamatatos, HIV vaccine design: The neutralizing antibody conundrum, Curr Opin Immunol, 24 (2012): 316-323. |
| [40] | [ L. Stamatatos,L. Morris,D. Burton,J. Mascola, Neutralizing antibodies generated during natural HIV-1 infection: Good news for an HIV-1 vaccine?, Nat Med, 15 (2009): 866-870. |
| [41] | [ L. Verkoczy,G. Kelsoe,M. Moody,B. Haynes, Role of immune mechanisms in induction of HIV-1 broadly neutralizing antibodies, Curr Opin Immunol, 23 (2011): 383-390. |
| [42] | [ G. Victora,L. Mesin, Clonal and cellular dynamics in germinal centers, Curr Opin Immunol, 28 (2014): 90-96. |
| [43] | [ C. Vinuesa, HIV and T follicular helper cells: A dangerous relationship, J Clin Invest, 122 (2012): 3059-3062. |
| [44] | [ C. Vinuesa,I. Sanz,M. Cook, Dysregulation of germinal centres in autoimmune disease, Nat Rev Immunol, 9 (2009): 845-857. |
| [45] | [ J. Weinstein,S. Hernandez,J. Craft, T cells that promote B-cell maturation in systemic autoimmunity, Immunol Rev, 247 (2012): 160-171. |
| [46] | [ I. Wollenberg,A. Agua-Doce,A. Hernandez,C. Almeida,V. Oliveira, Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells, J Immunol, 187 (2011): 4553-4560. |
| [47] | [ X. Wu,T. Zhou,J. Zhu,B. Zhang,I. Georgiev, Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing, Science, 333 (2011): 1593-1602. |
| [48] | [ X. Zhang,S. Ing,A. Fraser,M. Chen,O. Khan,J. Zakem, Follicular helper T cells: New insights into mechanisms of autoimmune diseases, Ochsner J, 13 (2013): 131-139. |
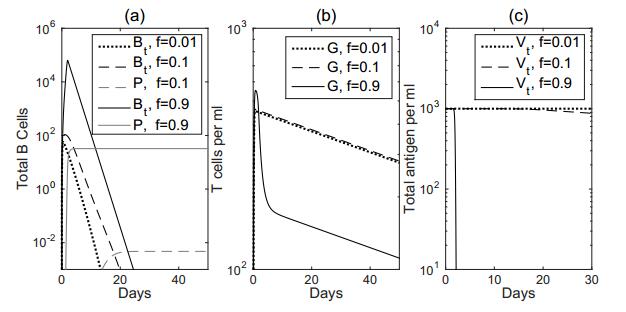
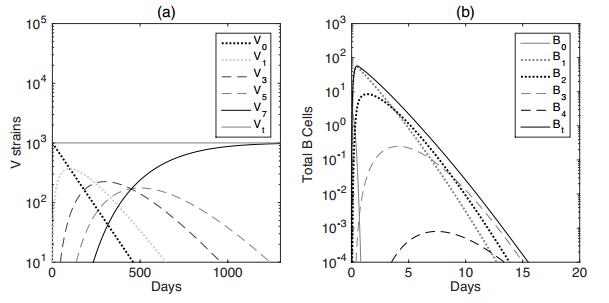
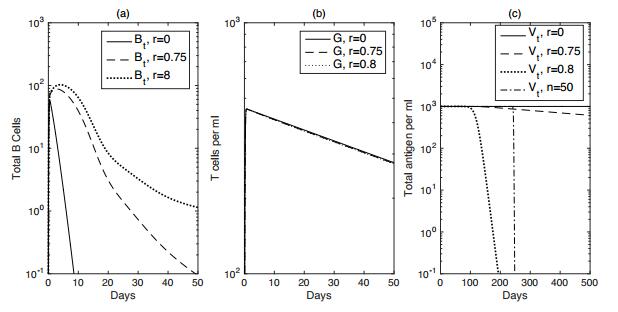
Figures(7) / Tables(2)

Samantha Erwin, Stanca M. Ciupe. Germinal center dynamics during acute and chronic infection[J]. Mathematical Biosciences and Engineering, 2017, 14(3): 655-671. doi: 10.3934/mbe.2017037







 DownLoad:
DownLoad: