Citation: Katrine O. Bangsgaard, Morten Andersen, Vibe Skov, Lasse Kjær, Hans C. Hasselbalch, Johnny T. Ottesen. Dynamics of competing heterogeneous clones in blood cancers explains multiple observations - a mathematical modeling approach[J]. Mathematical Biosciences and Engineering, 2020, 17(6): 7645-7670. doi: 10.3934/mbe.2020389

| [1] |
S. H. Orkin, L. I. Zon, Hematopoiesis: An evolving paradigm for stem cell biology, Cell, 132 (2008), 631-644. doi: 10.1016/j.cell.2008.01.025

|
| [2] |
S. N. Catlin, L. Busque, R. E. Gale, P. Guttorp, J. L. Abkowitz, The replication rate of human hematopoietic stem cells in vivo, Blood, 117 (2011), 4460-4466. doi: 10.1182/blood-2010-08-303537
|
| [3] |
H. Lee-Six, N. F. Øbro, M. S. Shepherd, S. Grossmann, K. Dawson, M. Belmonte, et al., Population dynamics of normal human blood inferred from somatic mutations, Nature, 561 (2018), 473-478. doi: 10.1038/s41586-018-0497-0
|
| [4] | D. Dingli, A. Traulsen, J. M. Pacheco, Compartmental architecture and dynamics of hematopoiesis (architecture of hematopoiesis), PLoS ONE, 2 (2007), e345. |
| [5] |
H. Vaziri, W. Dragowska, R. C. Allsopp, T. E. Thomas, C. B. Harley, P. M. Lansdorp, Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age, Proc. Natl. Sci. Acad., 91 (1994), 9857-9860. doi: 10.1073/pnas.91.21.9857
|
| [6] |
S. Y. Chen, Y. C. Huang, S. P. Liu, F. J. Tsai, W. C. Shyu, S. Z. Lin, An overview of concepts for cancer stem cells, Cell Trans., 20 (2011), 113-120. doi: 10.3727/096368910X532837
|
| [7] | A. Tefferi, P. Guglielmelli, D. R. Larson, C. Finke, E. A. Wassie, L. Pieri, et al., Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis, Blood, 124 (2014), 2507-2513. |
| [8] | J. M. Shammo, B. L. Stein, Mutations in MPNs: prognostic implications, window to biology, and impact on treatment decisions, Hematology, 2016 (2016), 552-560. |
| [9] |
P. J. Campbell, A. R. Green, The myeloproliferative disorders, N. Engl. J. Med., 355 (2006), 2452-2466. doi: 10.1056/NEJMra063728
|
| [10] |
L. A. Anderson, M. F. McMullin, Epidemiology of MPN: What do we know?, Curr. Hematol. Malig. Rep., 9 (2014), 340-349. doi: 10.1007/s11899-014-0228-z
|
| [11] | H. C. Hasselbalch, M. E. Bjørn, MPNs as inflammatory diseases: The evidence, consequences, and perspectives, Mediators Inflammation, 2015 (2015), 1-16. |
| [12] |
S. Y. Kristinsson, O. Landgren, J. Samuelsson, M. Bjorkholm, L. R. Goldin, Autoimmunity and the risk of myeloproliferative neoplasms, Haematologica, 95 (2010), 1216-1220. doi: 10.3324/haematol.2009.020412
|
| [13] |
R. Marchioli, G. Finazzi, R. Landolfi, J. Kutti, H. Gisslinger, C. Patrono, et al., Vascular and neoplastic risk in a large cohort of patients with polycythemia vera, J. Clin. Onco., 23 (2005), 2224-2232. doi: 10.1200/JCO.2005.07.062
|
| [14] | G. Multhoff, M. Molls, J. Radons, Chronic inflammation in cancer development, Front. Immunol., 2 (2012). |
| [15] |
J. Todoric, L. Antonucci, M. Karin, Targeting inflammation in cancer prevention and therapy, Cancer Prev. Res., 9 (2016), 895-905. doi: 10.1158/1940-6207.CAPR-16-0209
|
| [16] |
G. J. Titmarsh, A. S. Duncombe, M. F. McMullin, M. O'Rorke, R. Mesa, F. D. Vocht, et al., How common are myeloproliferative neoplasms? A systematic review and meta-analysis, Am. J. Hematol., 89 (2014), 581-587. doi: 10.1002/ajh.23690
|
| [17] | M. Andersen, Z. Sajid, R. K. Pedersen, J. Gudmand-Hoeyer, C. Ellervik, V. Skov, et al., Mathematical modelling as a proof of concept for MPNs as a human inflammation model for cancer development, PLOS ONE, 12 (2017), e0183620. |
| [18] |
Z. Sajid, M. Andersen, J. T. Ottesen, Mathematical analysis of the cancitis model and the role of inflammation in blood cancer progression, Math. Biosci. Eng., 16 (2019), 8268-8289. doi: 10.3934/mbe.2019418
|
| [19] | M. Andersen, H. Hasselbalch, L. Kjær, V. Skov, J. Ottesen, Global dynamics of healthy and cancer cells competing in the hematopoietic system, Math. Biosci., 2020 (2020), 108372. |
| [20] |
J. T. Ottesen, R. K. Pedersen, Z. Sajid, J. Gudmand-Hoeyer, K. O. Bangsgaard, V. Skov, et al., Bridging blood cancers and inflammation: The reduced cancitis model, J. Theor. Biol., 465 (2019), 90-108. doi: 10.1016/j.jtbi.2019.01.001
|
| [21] |
J. Ottesen, R. Pedersen, M. Dam, T. Knudsen, V. Skov, L. Kjær, et al., Mathematical modeling of MPNs offers understanding and decision support for personalized treatment, Cancers, 12 (2020), 2119. doi: 10.3390/cancers12082119
|
| [22] |
B. J. Kennedy, Cyclic leukocyte oscillations in chronic myelogenous leukemia during hydroxyurea therapy, Blood, 35 (1970), 751-760. doi: 10.1182/blood.V35.6.751.751
|
| [23] |
G. Chikkappa, G. Borner, H. Burlington, A. Chanana, E. Cronkite, S. Ohl, et al., Periodic oscillation of blood leukocytes, platelets, and reticulocytes in a patient with chronic myelocytic leukemia, Blood, 47 (1976), 1023-1030. doi: 10.1182/blood.V47.6.1023.1023
|
| [24] |
P. Fortin, M. C. Mackey, Periodic chronic myelogenous leukaemia: spectral analysis of blood cell counts and aetiological implications, Br. J. Haematol., 104 (1999), 336-345. doi: 10.1046/j.1365-2141.1999.01168.x
|
| [25] |
Y. Hirayama, S. Sakamaki, Y. Tsuji, T. Matsunaga, Y. Niitsu, Cyclic platelet and leukocyte count oscillation in chronic myelocytic leukemia regulated by the negative feedback of transforming growth factor beta, Int. J. Hematol., 77 (2003), 71-74. doi: 10.1007/BF02982605
|
| [26] | A. Besse, G. D. Clapp, S. Bernard, F. E. Nicolini, D. Levy, T. Lepoutre, Stability analysis of a model of interaction between the immune system and cancer cells in chronic myelogenous leukemia, Bull. Math. Biol., 80 (2017), 1084-1110. |
| [27] |
G. D. Clapp, T. Lepoutre, R. E. Cheikh, S. Bernard, J. Ruby, H. Labussiere-Wallet, et al., Implication of the autologous immune system in BCR-ABL transcript variations in chronic myelogenous leukemia patients treated with imatinib, Cancer Res., 75 (2015), 4053-4062. doi: 10.1158/0008-5472.CAN-15-0611
|
| [28] | F. Knauer, T. Stiehl, A. Marciniak-Czochra, Oscillations in a white blood cell production model with multiple differentiation stages, J. Math. Biol., 80 (2020), 575-600. |
| [29] | J. H. Baird, C. P. Minniti, J.-M. Lee, X. Tian, C. Wu, M. Jackson, et al., Oscillatory haematopoiesis in adults with sickle cell disease treated with hydroxycarbamide, Br. J. Hematol., 168 (2014), 737-746. |
| [30] |
J. Tauscher, F. Siegel, P. E. Petrides, Hydroxyurea induced oscillations in twelve patients with polycythemia vera, Haematologica, 95 (2010), 1227-1229. doi: 10.3324/haematol.2010.022178
|
| [31] |
A. Tefferi, M. A. Elliott, P. C. Kao, S. Yoon, I. El-Hemaidi, T. C. Pearson, Hydroxyurea-induced marked oscillations of platelet counts in patients with polycythemia vera, Blood, 96 (2000), 1582-1584. doi: 10.1182/blood.V96.4.1582
|
| [32] |
S. Jaiswal, P. Fontanillas, J. Flannick, A. Manning, P. V. Grauman, B. G. Mar, et al., Age-related clonal hematopoiesis associated with adverse outcomes, N. Engl. J. Med., 371 (2014), 2488-2498. doi: 10.1056/NEJMoa1408617
|
| [33] | M. Heuser, F. Thol, A. Ganser, Clonal hematopoiesis of indeterminate potential, Dtsch. Ärzteblatt Int., 113 (2016), 317. |
| [34] | D. S. Park, A. A. Akuffo, D. E. Muench, H. L. Grimes, P. K. Epling-Burnette, P. K. Maini, et al., Clonal hematopoiesis of indeterminate potential and its impact on patient trajectories after stem cell transplantation, PLoS Comput. Biol., 15 (2019), e1006913. |
| [35] | J. Cortes, M. O'Dwyer, Clonal evolution in chronic myelogenous leukemia, Haematol./Oncol. Clin. North Am., 18 (2004), 671-684. |
| [36] | A. Schuh, J. Becq, S. Humphray, A. Alexa, A. Burns, R. Clifford, et al., Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns, Blood, 120 (2012), 4191-4196. |
| [37] |
M. J. Walter, D. Shen, L. Ding, J. Shao, D. C. Koboldt, K. Chen, et al., Clonal architecture of secondary acute myeloid leukemia, N. Engl. J. Med., 366 (2012), 1090-1098. doi: 10.1056/NEJMoa1106968
|
| [38] |
L. Ding, T. J. Ley, D. E. Larson, C. A. Miller, D. C. Koboldt, J. S. Welch, et al., Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing, Nature, 481 (2012), 506-510. doi: 10.1038/nature10738
|
| [39] | K. H. Allison, G. W. Sledge, Heterogeneity and cancer, Oncology, 28 (2014), A11. |
| [40] | T. Stiehl, N. Baran, A. D. Ho, A. Marciniak-Czochra, Clonal selection and therapy resistance in acute leukaemias: mathematical modelling explains different proliferation patterns at diagnosis and relapse, J. R. Soc. Interface, 11 (2014), 20140079. |
| [41] |
G. P. Dunn, L. J. Old, R. D. Schreiber, The three es of cancer immunoediting, Ann. Rev. Immunol., 22 (2004), 329-360. doi: 10.1146/annurev.immunol.22.012703.104803
|
| [42] | R. J. Jones, S. A. Armstrong, Cancer stem cells in hematopoietic malignancies, Biol. Blood Marrow Trans., 14 (2008), 12-16. |
| [43] |
A. J. Mead, A. Mullally, Myeloproliferative neoplasm stem cells, Blood, 129 (2017), 1607-1616. doi: 10.1182/blood-2016-10-696005
|
| [44] | B. Craver, K. Alaoui, R. Scherber, A. Fleischman, The critical role of inflammation in the pathogenesis and progression of myeloid malignancies, Cancers, 10 (2018), 2-18. |
| [45] |
L. Shlush, A. Mitchell, L. Heisler, S. Abelson, A. Trotman-Grant, J. Medeiros, et al., Tracing the origins of relapse in acute mueloid leukemia to stem cells, Nature, 547 (2017), 104-108. doi: 10.1038/nature22993
|
| [46] | B. Jonas, On the origin of relapse in AML, Sci. Transl. Med., 9(398) (2017), 1-2. |
| [47] | B. Jonas, Stem cells make leukemia grow again, EMBO J., 36(18) (2017), 2667-2669. |
| [48] |
L. MacPherson, M. Dawson, Survival of the fittest: Darwinian selection underpines chemotherapy resistance in AML, Cell Stem Cell, 21 (2017), 291-292. doi: 10.1016/j.stem.2017.08.004
|
| [49] | J. Hofbauer, J.-H. So, Multiple limit cycles for three dimensional Lotka-Volterra equations, Appl. Math. Lett., 7 (1994), 65-70. |
| [50] | M. L. Zeeman, Hopf bifurcations in competitive three-dimensional Lotka-Volterra systems, Dyn. Stab. Syst., 8 (1993), 189-216. |
| [51] | M. L. Zeeman, P. Van Den Driessche, Three-dimensional competitive Lotka-Volterra systems with no periodic orbits, SIAM J. Appl. Math., 58 (1998), 227-234. |
| [52] |
R. M. May, W. J. Leonard, Nonlinear aspects of competition between three species, SIAM J. Appl. Math., 29 (1975), 243-253. doi: 10.1137/0129022
|
| [53] |
M. E. Gilpin, Limit cycles in competition communities, Am. Nat., 109 (1975), 51-60. doi: 10.1086/282973
|
| [54] |
J. Huisman, F. J. Weissing, Biological conditions for oscillations and chaos generated by multi-species competition, Ecology, 82 (2001), 2682-2695. doi: 10.1890/0012-9658(2001)082[2682:BCFOAC]2.0.CO;2
|
| [55] | J. D. Murray, Interdisciplinary applied mathematics, in Mathematical biology, 3rd edition, Springer, New York, 2003. |
| [56] |
R. K. Pedersen, T. A. Knudsen, Z. Sajid, J. Gudmand-Hoeyer, V. Skov, L. Kjær, et al., Data-driven analysis of JAK2V617F kinetics during interferon-alpha2 treatment of patients with polycythemia vera and related neoplasms, Cancer Med., 9 (2020), 2039-2051. doi: 10.1002/cam4.2741
|
 MBE-17-06-389-supplementary.pdf MBE-17-06-389-supplementary.pdf |
 |
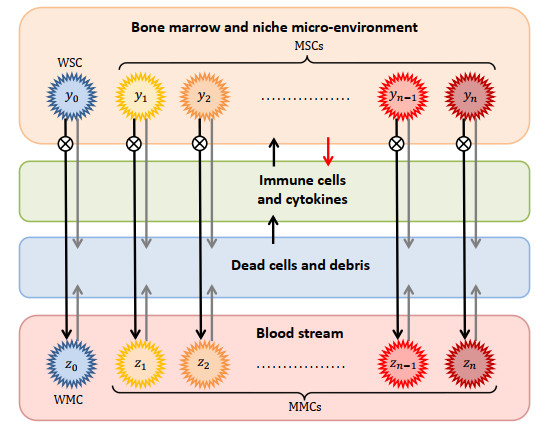
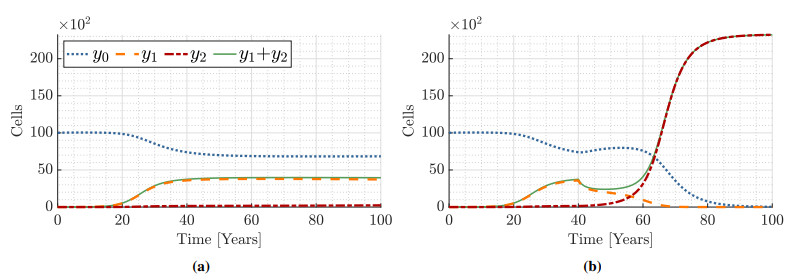
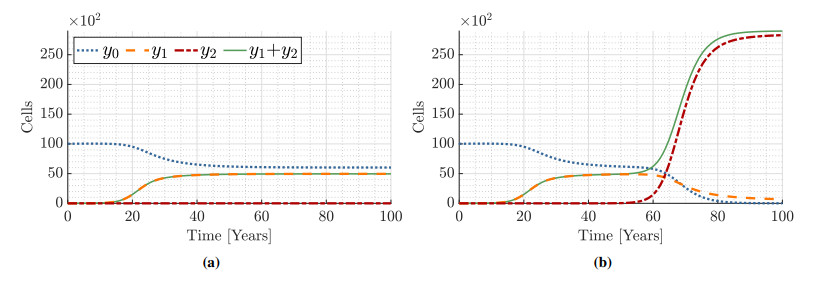
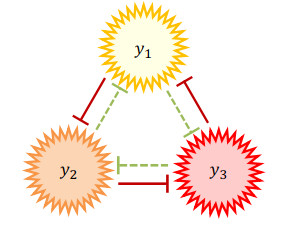
Figures(9) / Tables(15)

Katrine O. Bangsgaard, Morten Andersen, Vibe Skov, Lasse Kjær, Hans C. Hasselbalch, Johnny T. Ottesen. Dynamics of competing heterogeneous clones in blood cancers explains multiple observations - a mathematical modeling approach[J]. Mathematical Biosciences and Engineering, 2020, 17(6): 7645-7670. doi: 10.3934/mbe.2020389







 DownLoad:
DownLoad: