Citation: Xiangmei Li, Kamran, Absar Ul Haq, Xiujun Zhang. Numerical solution of the linear time fractional Klein-Gordon equation using transform based localized RBF method and quadrature[J]. AIMS Mathematics, 2020, 5(5): 5287-5308. doi: 10.3934/math.2020339

| [1] |
F. Yousef, M. Alquran, I. Jaradat, et al. Ternary-fractional differential transform schema: Theory and application, Adv. Differ. Equ., 2019 (2019), 1-13. doi: 10.1186/s13662-018-1939-6

|
| [2] | S. Bhatter, A. Mathur, D. Kumar, et al. A new analysis of fractional Drinfeld-Sokolov-Wilson model with exponential memory, Physica A, 537 (2020), 122578. |
| [3] |
A. Goswami, J. Singh, D. Kumar, et al. An efficient analytical approach for fractional equal width equations describing hydro-magnetic waves in cold plasma, Physica A, 524 (2019), 563-575. doi: 10.1016/j.physa.2019.04.058
|
| [4] | D. Kumar, F. Tchier, J. Singh, et al. An efficient computational technique for fractal vehicular traffic flow, Entropy, 20 (2018), 1-9. |
| [5] |
D. Kumar, J. Singh, D. Baleanu, On the analysis of vibration equation involving a fractional derivative with Mittag-Leffler law, Math. Meth. Appl. Sci., 43 (2020), 443-457. doi: 10.1002/mma.5903
|
| [6] |
A. Yusuf, M. Inc, A. I. Aliyu, et al. Efficiency of the new fractional derivative with nonsingular Mittag-Leffler kernel to some nonlinear partial differential equations, Chaos Soliton Fract., 116 (2018), 220-226. doi: 10.1016/j.chaos.2018.09.036
|
| [7] |
D. Kumar, J. Singh, K. Tanwar, et al. A new fractional exothermic reactions model having constant heat source in porous media with power, exponential and Mittag-Leffler laws, Int. J. Heat Mass Tran., 138 (2019), 1222-1227. doi: 10.1016/j.ijheatmasstransfer.2019.04.094
|
| [8] | D. Kumar, J. Singh, S. D. Purohit, et al. A hybrid analytical algorithm for nonlinear fractional wave-like equations, Math. Model. Nat. Pheno., 14 (2019), 1-13. |
| [9] | M. J. Ablowitz, M. A. Ablowitz, P. A. Clarkson, et al. Solitons, Nonlinear Evolution Equations and Inverse Scatterin, Cambridge university press, 1991. |
| [10] | A. M. Yang, Y. Z. Zhang, C. Cattani, et al. Application of local fractional series expansion method to solve Klein-Gordon equations on Cantor sets, Abstr. Appl. Anal., 2014 (2014), 1-6. |
| [11] |
A. M. Wazwaz, New travelling wave solutions to the Boussinesq and the Klein-Gordon equations, Commun. Nonlinear Sci., 13 (2008), 889-901. doi: 10.1016/j.cnsns.2006.08.005
|
| [12] | A. M. Wazwaz, The tanh and the sine-cosine methods for compact and noncompact solutions of the nonlinear Klein-Gordon equation, Appl. Math. Comput., 167 (2005), 1179-1195. |
| [13] | A. S. V. R. Kanth, K. Aruna, Differential transform method for solving the linear and nonlinear Klein-Gordon equation, Comput. Phys. Commun., 180 (2018), 708-711. |
| [14] |
K. Hosseini, P. Mayeli, R. Ansar, Modified Kudryashov method for solving the conformable timefractional Klein-Gordon equations with quadratic and cubic nonlinearities, Optik, 130 (2017), 737-742. doi: 10.1016/j.ijleo.2016.10.136
|
| [15] |
K. Hosseini, P. Mayeli, D. Kuma, New exact solutions of the coupled sine-Gordon equations in nonlinear optics using the modified Kudryashov method, J. Mod. Optic., 65 (2018), 361-364. doi: 10.1080/09500340.2017.1380857
|
| [16] |
K. Hosseini, P. Mayeli, R. Ansar, Bright and singular soliton solutions of the conformable timefractional Klein-Gordon equations with different nonlinearities, Wave. Random Complex, 28 (2018), 426-434. doi: 10.1080/17455030.2017.1362133
|
| [17] | K. Hosseini, Y. J. Xu, P. Mayeli, et al. A study on the conformable time-fractional Klein-Gordon equations with quadratic and cubic nonlinearitiesA study on the conformable time-fractional Klein-Gordon equations with quadratic and cubic nonlinearities, Optoelectron. Adv. Mat., 11 (2017), 423-429. |
| [18] |
E. Yusufoğlu, The variational iteration method for studying the Klein-Gordon equation, Appl. Math. Lett., 21 (2008), 669-674. doi: 10.1016/j.aml.2007.07.023
|
| [19] | M. Alaroud, M. Al-Smadi, O. A. Arqub, et al. Numerical Solutions of Linear Time-fractional Klein-Gordon Equation by Using Power Series Approach, SSRN Electron. J., 2018 (2018), 1-6. |
| [20] |
M. Kurulay, Solving the fractional nonlinear Klein-Gordon equation by means of the homotopy analysis method, Adv. Differ, Equ., 2012 (2012), 1-8. doi: 10.1186/1687-1847-2012-1
|
| [21] | K. A. Gepreel, M. S. Mohamed, Analytical approximate solution for nonlinear space-time fractional Klein-Gordon equation, Chinese Phys. B, 22 (2013), 010201. |
| [22] |
A. K. Golmankhaneh, A. K. Golmankhaneh, D. Baleanu, On nonlinear fractional Klein-Gordon equation, Signal Process., 91 (2011), 446-451. doi: 10.1016/j.sigpro.2010.04.016
|
| [23] | E. A. B. Abdel-Salam, E. A. Yousif, Solution of nonlinear space-time fractional differential equations using the fractional Riccati expansion method, Math. Probl. Eng., 2013 (2013), 1-6. |
| [24] |
D. Kumar, J. Singh, D. Baleanu, A hybrid computational approach for Klein-Gordon equations on Cantor sets, Nonlinear Dynam., 87 (2017), 511-517. doi: 10.1007/s11071-016-3057-x
|
| [25] |
A. Mohebbi, M. Abbaszadeh, M. Dehghan, High-order difference scheme for the solution of linear time fractional Klein-Gordon equations, Numer. Meth. Part. Differ. Equ., 30 (2014), 1234-1253. doi: 10.1002/num.21867
|
| [26] |
M. Dehghan, M. Abbaszadeh, A. Mohebbi, An implicit RBF meshless approach for solving the time fractional nonlinear sine-Gordon and Klein-Gordon equations, Eng. Anal. Bound. Elem., 50 (2015), 412-434. doi: 10.1016/j.enganabound.2014.09.008
|
| [27] |
M. M. Khader, An efficient approximate method for solving linear fractional Klein-Gordon equation based on the generalized Laguerre polynomials, Int. J. Comput. Math., 90 (2013), 1853-1864. doi: 10.1080/00207160.2013.764994
|
| [28] | G. Hariharan, Wavelet method for a class of fractional Klein-Gordon equations, J. Comput. Nonlinear Dynam., 8 (2013), 1-6. |
| [29] | H. Singh, D. Kumar, J. Singh, et al. A reliable numerical algorithm for the fractional klein-gordon equation, Eng. Trans., 67 (2019), 21-34. |
| [30] | S. Vong, Z. Wang, A compact difference scheme for a two dimensional fractional Klein-Gordon equation with Neumann boundary conditions, Entropy, 274 (2014), 268-282. |
| [31] |
M. Dehghan, A. Shokri, Numerical solution of the nonlinear Klein-Gordon equation using radial basis functions, J. Comput. Appl. Math., 230 (2009), 400-410. doi: 10.1016/j.cam.2008.12.011
|
| [32] | M. Dehghan, A. Shokri, A method of solution for certain problems of transient heat conduction, AIAA J., 8 (1968), 2004-2009. |
| [33] |
G. J. Moridis, D. L. Reddell, The Laplace transform finite difference method for simulation of flow through porous media, Water Resour. Res., 27 (1991), 1873-1884. doi: 10.1029/91WR01190
|
| [34] | G. J. Moridis, D. L. Reddell, The Laplace transform boundary element (LTBE) method for the solution of diffusion-type equations, In: Boundary Elements XIII, Springer, Dordrecht, 1991, 83-97. |
| [35] | G. J. Moridis, D. L. Reddell, The Laplace transform finite element (LTFE) numerical method for the solution of the ground water equations, paper H22C-4, AGU 91 Spring Meeting, Baltimore, May 28-31, 1991, EOS Trans. of the AGU, 72 (1991), 17. |
| [36] |
E. A. Sudicky, R. G. McLaren, The Laplace Transform Galerkin Technique for large-scale simulation of mass transport in discretely fractured porous formations, Water Resour. Res., 28 (1992), 499-514. doi: 10.1029/91WR02560
|
| [37] | G. J. Moridis, E. J. Kansa, The Laplace transform multiquadric method: A highly accurate scheme for the numerical solution of partial differentia, J. Appl. Sci. Comput., (1993), 55910181. |
| [38] |
Q. T. L. Gia, W. Mclean, Solving the heat equation on the unit sphere via Laplace transforms and radial basis functions, Adv. Comput. Math., 40 (2014), 353-375. doi: 10.1007/s10444-013-9311-6
|
| [39] |
W. McLean, V. Thomee, Time discretization of an evolution equation via Laplace transforms, IMA J. Numer. Anal., 24 (2004), 439-463. doi: 10.1093/imanum/24.3.439
|
| [40] |
M. L. Fernandez, C. Palencia, On the numerical inversion of the Laplace transform of certain holomorphic mappings, Appl. Numer. Math., 51 (2004), 289-303. doi: 10.1016/j.apnum.2004.06.015
|
| [41] |
W. McLean, V. Thomee, Numerical solution via Laplace transforms of a fractional order evolution equation, J. Integral Equ. Appl., 22 (2010), 57-94. doi: 10.1216/JIE-2010-22-1-57
|
| [42] |
J. A. C. Weideman, L. N. Trefethen, Parabolic and Hyperbolic contours for computing the Bromwich integral, Math. Comput., 76 (2007), 1341-1356. doi: 10.1090/S0025-5718-07-01945-X
|
| [43] |
D. Sheen, I. H. Shaon, V. Thomee, A parallel method for time discretization of parabolic equations based on Laplace transformation and quadrature, IMA J. Numer.l Anal., 23 (2003), 269-299. doi: 10.1093/imanum/23.2.269
|
| [44] |
Z. J. Fu, W. Chen, H. T. Yang, Boundary particle method for Laplace transformed time fractional diffusion equations, J. Comput. Phys., 235 (2013), 52-66. doi: 10.1016/j.jcp.2012.10.018
|
| [45] |
M. Uddin, A. Ali, On the approximation of time-fractional telegraph equations using localized kernel-based method, Adv. Differ. Equ., 2018 (2018), 1-14. doi: 10.1186/s13662-017-1452-3
|
| [46] |
M. Uddin, A. Ali, A localized transform-based meshless method for solving time fractional wave- diffusion equation, Eng. Anal. Bound. Elem., 92 (2018), 108-113. doi: 10.1016/j.enganabound.2017.10.021
|
| [47] | K. B. Oldham, J. Spanier, The fractional calculus theory and applications of differentiation and integration to arbitrary order, Academic Press, New York, London, 1974. |
| [48] |
M. Uddin, On the selection of a good value of shape parameter in solving time dependent partial differential equations using RBF approximation method, Appl. Math. Model., 38 (2014), 135-144. doi: 10.1016/j.apm.2013.05.060
|
| [49] | R. E. Carlson, T. A. Foley, The parameter r2 in multiquadric interpolation, Comput. Math. Appl., 21 ((1991), 29-42. |
| [50] | R. E. Carlson, T. A. Foley, Near optimal parameter selection for multiquadric interpolation, Manuscript, Computer Science and Engineering Department, Arizona State University, Tempe., 20 (1994). |
| [51] |
D. Kumar, F. Tchier, J. Singh, et al. Error estimates and condition numbers for radial basis function interpolation, Adv. Comput. Math., 3 (1995), 251-264. doi: 10.1007/BF02432002
|
| [52] | L. N. Trefethen, D. Bau, Numerical Linear Algebra, SIAM, 1997. |
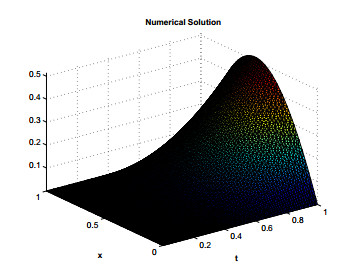
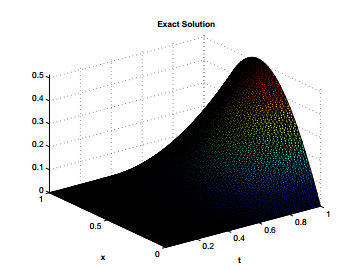
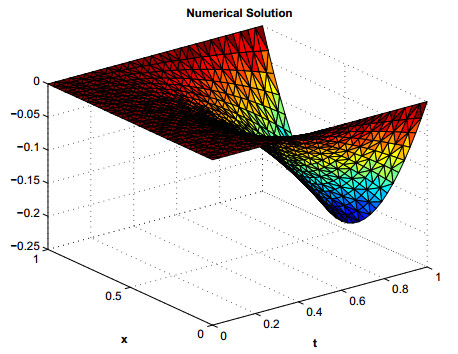
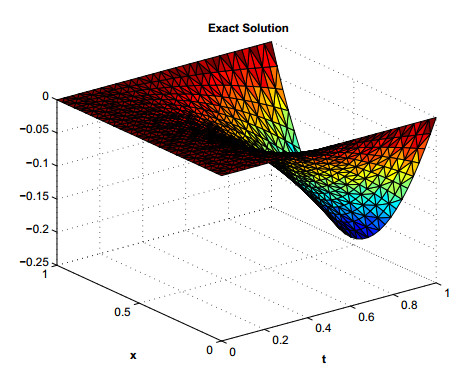
Figures(12) / Tables(7)

Xiangmei Li, Kamran, Absar Ul Haq, Xiujun Zhang. Numerical solution of the linear time fractional Klein-Gordon equation using transform based localized RBF method and quadrature[J]. AIMS Mathematics, 2020, 5(5): 5287-5308. doi: 10.3934/math.2020339







 DownLoad:
DownLoad: