The deregulated genetic factors are critically associated with idiopathic pulmonary arterial hypertension (IPAH) development and progression. However, the identification of hub-transcription factors (TFs) and miRNA-hub-TFs co-regulatory network-mediated pathogenesis in IPAH remains lacking.
We used GSE48149, GSE113439, GSE117261, GSE33463, and GSE67597 for identifying key genes and miRNAs in IPAH. We used a series of bioinformatics approaches, including R packages, protein-protein interaction (PPI) network, and gene set enrichment analysis (GSEA) to identify the hub-TFs and miRNA-hub-TFs co-regulatory networks in IPAH. Also, we employed a molecular docking approach to evaluate the potential protein-drug interactions.
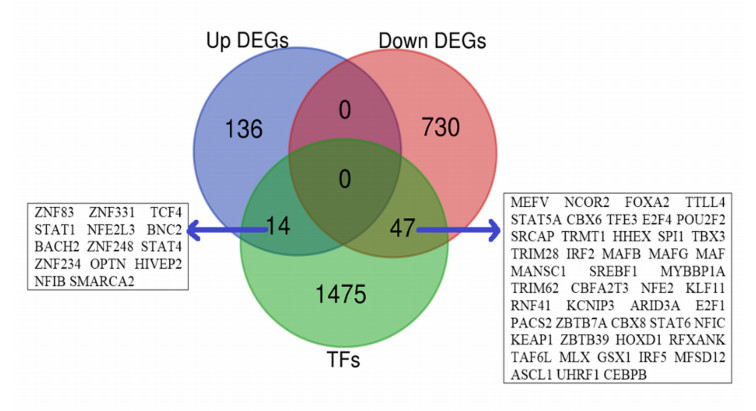
We found that 14 TFs encoding genes, including ZNF83, STAT1, NFE2L3, and SMARCA2 are upregulated, and 47 TFs encoding genes, including NCOR2, FOXA2, NFE2, and IRF5 are downregulated in IPAH relative to the control. Then, we identified the differentially expressed 22 hub-TFs encoding genes, including four upregulated (STAT1, OPTN, STAT4, and SMARCA2) and 18 downregulated (such as NCOR2, IRF5, IRF2, MAFB, MAFG, and MAF) TFs encoding genes in IPAH. The deregulated hub-TFs regulate the immune system, cellular transcriptional signaling, and cell cycle regulatory pathways. Moreover, the identified differentially expressed miRNAs (DEmiRs) are involved in the co-regulatory network with hub-TFs. The six hub-TFs encoding genes, including STAT1, MAF, CEBPB, MAFB, NCOR2, and MAFG are consistently differentially expressed in the peripheral blood mononuclear cells of IPAH patients, and these hub-TFs showed significant diagnostic efficacy in distinguishing IPAH cases from the healthy individuals. Moreover, we revealed that the co-regulatory hub-TFs encoding genes are correlated with the infiltrations of various immune signatures, including CD4 regulatory T cells, immature B cells, macrophages, MDSCs, monocytes, Tfh cells, and Th1 cells. Finally, we discovered that the protein product of STAT1 and NCOR2 interacts with several drugs with appropriate binding affinity.
The identification of hub-TFs and miRNA-hub-TFs co-regulatory networks may provide a new avenue into the mechanism of IPAH development and pathogenesis.
Citation: Qian Li, Minawaer Hujiaaihemaiti, Jie Wang, Md. Nazim Uddin, Ming-Yuan Li, Alidan Aierken, Yun Wu. Identifying key transcription factors and miRNAs coregulatory networks associated with immune infiltrations and drug interactions in idiopathic pulmonary arterial hypertension[J]. Mathematical Biosciences and Engineering, 2023, 20(2): 4153-4177. doi: 10.3934/mbe.2023194
The deregulated genetic factors are critically associated with idiopathic pulmonary arterial hypertension (IPAH) development and progression. However, the identification of hub-transcription factors (TFs) and miRNA-hub-TFs co-regulatory network-mediated pathogenesis in IPAH remains lacking.
We used GSE48149, GSE113439, GSE117261, GSE33463, and GSE67597 for identifying key genes and miRNAs in IPAH. We used a series of bioinformatics approaches, including R packages, protein-protein interaction (PPI) network, and gene set enrichment analysis (GSEA) to identify the hub-TFs and miRNA-hub-TFs co-regulatory networks in IPAH. Also, we employed a molecular docking approach to evaluate the potential protein-drug interactions.
We found that 14 TFs encoding genes, including ZNF83, STAT1, NFE2L3, and SMARCA2 are upregulated, and 47 TFs encoding genes, including NCOR2, FOXA2, NFE2, and IRF5 are downregulated in IPAH relative to the control. Then, we identified the differentially expressed 22 hub-TFs encoding genes, including four upregulated (STAT1, OPTN, STAT4, and SMARCA2) and 18 downregulated (such as NCOR2, IRF5, IRF2, MAFB, MAFG, and MAF) TFs encoding genes in IPAH. The deregulated hub-TFs regulate the immune system, cellular transcriptional signaling, and cell cycle regulatory pathways. Moreover, the identified differentially expressed miRNAs (DEmiRs) are involved in the co-regulatory network with hub-TFs. The six hub-TFs encoding genes, including STAT1, MAF, CEBPB, MAFB, NCOR2, and MAFG are consistently differentially expressed in the peripheral blood mononuclear cells of IPAH patients, and these hub-TFs showed significant diagnostic efficacy in distinguishing IPAH cases from the healthy individuals. Moreover, we revealed that the co-regulatory hub-TFs encoding genes are correlated with the infiltrations of various immune signatures, including CD4 regulatory T cells, immature B cells, macrophages, MDSCs, monocytes, Tfh cells, and Th1 cells. Finally, we discovered that the protein product of STAT1 and NCOR2 interacts with several drugs with appropriate binding affinity.
The identification of hub-TFs and miRNA-hub-TFs co-regulatory networks may provide a new avenue into the mechanism of IPAH development and pathogenesis.

| [1] |
M. M. Hoeper, M. Humbert, R. Souza, M. Idrees, S. M. Kawut, K. Sliwa-Hahnle, et al., A global view of pulmonary hypertension, Lancet Respir. Med., 4 (2016), 306–322. https://doi.org/10.1016/s2213-2600(15)00543-3 doi: 10.1016/s2213-2600(15)00543-3

|
| [2] |
H. Zeng, X. Liu, Y. Zhang, Identification of potential biomarkers and immune infiltration characteristics in idiopathic pulmonary arterial hypertension using bioinformatics analysis, Front. Cardiovasc. Med., 8 (2021). https://doi.org/10.3389/fcvm.2021.624714 doi: 10.3389/fcvm.2021.624714
|
| [3] |
N. Galiè, M. Humbert, J. Vachiery, S. Gibbs, I. Lang, A. Torbicki, et al., 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension, Rev. Esp. Cardiol. (Engl. Ed.), 69 (2016), 177. https://doi.org/10.1016/j.rec.2016.01.002 doi: 10.1016/j.rec.2016.01.002
|
| [4] |
V. V. McLaughlin, M. D. McGoon, Pulmonary Arterial Hypertension, Circulation, 114 (2006), 1417–1431. https://doi.org/10.1161/CIRCULATIONAHA.104.503540 doi: 10.1161/CIRCULATIONAHA.104.503540
|
| [5] | P. Pahal, S. Sharma, Idiopathic Pulmonary Artery Hypertension, StatPearls Publishing, Florida, 2022. |
| [6] |
E. Spiekerkoetter, S. M. Kawut, V. A. de Jesus Perez, New and emerging therapies for pulmonary arterial hypertension, Annu. Rev. Med., 70 (2019), 45–59. https://doi.org/10.1146/annurev-med-041717-085955 doi: 10.1146/annurev-med-041717-085955
|
| [7] |
J. Y. Cao, K. M. Wales, R. Cordina, E. M. T. Lau, D. S. Celermajer, Pulmonary vasodilator therapies are of no benefit in pulmonary hypertension due to left heart disease: A meta-analysis, Int. J. Cardiol., 273 (2018), 213–220. https://doi.org/10.1016/j.ijcard.2018.09.043 doi: 10.1016/j.ijcard.2018.09.043
|
| [8] |
L. Yan, Q. Luo, Z. Zhao, Q. Zhao, Q. Jin, Y. Zhang, et al., Nocturnal hypoxia in patients with idiopathic pulmonary arterial hypertension, Pulm. Circ., 10 (2020), 1–7. https://doi.org/10.1177/2045894019885364 doi: 10.1177/2045894019885364
|
| [9] |
N. W. Morrell, M. A. Aldred, W. K. Chung, C. G. Elliott, W. C. Nichols, F. Soubrier, et al., Genetics and genomics of pulmonary arterial hypertension, Eur. Respir. J., 53 (2019), 1801899. https://doi.org/10.1183/13993003.01899-2018 doi: 10.1183/13993003.01899-2018
|
| [10] |
M. A. Aldred, J. Vijayakrishnan, V. James, F. Soubrier, M. A. Gomez-Sanchez, G. Martensson, et al., BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension, Hum. Mutat., 27 (2006), 212–213. https://doi.org/10.1002/humu.9398 doi: 10.1002/humu.9398
|
| [11] |
S. H. Choi, Y. Jung, J. Jang, S. Han, Idiopathic pulmonary arterial hypertension associated with a novel frameshift mutation in the bone morphogenetic protein receptor Ⅱ gene and enhanced bone morphogenetic protein signaling, Medicine, 98 (2019), e17594. https://doi.org/10.1097/MD.0000000000017594 doi: 10.1097/MD.0000000000017594
|
| [12] |
A. Chida, M. Shintani, T. Nakayama, Y. Furutani, E. Hayama, K. Inai, et al., Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension, Circ. J., 76 (2012), 1501–1508. https://doi.org/10.1253/circj.cj-11-1281 doi: 10.1253/circj.cj-11-1281
|
| [13] |
D. Saygin, T. Tabib, H. E. T. Bittar, E. Valenzi, J. Sembrat, S. Y. Chan, et al., Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension, Pulm. Circ., 10 (2020), 1–15. https://doi.org/10.1177/2045894020908782 doi: 10.1177/2045894020908782
|
| [14] |
Y. Wu, J. Wharton, R. Walters, E. Vasilaki, J. Aman, L. Zhao, et al., The pathophysiological role of novel pulmonary arterial hypertension gene SOX17, Eur. Respir. J., (2021). https://doi.org/10.1183/13993003.04172-2020 doi: 10.1183/13993003.04172-2020
|
| [15] |
C. S. Park, S. H. Kim, H. Y. Yang, J. Kim, R. T. Schermuly, Y. S. Cho, et al., Sox17 deficiency promotes pulmonary arterial hypertension via HGF/c-Met signaling, Circ. Res., 131 (2022), 792–806. https://doi.org/10.1161/CIRCRESAHA.122.320845 doi: 10.1161/CIRCRESAHA.122.320845
|
| [16] |
T. Wang, S. Wang, Y. Xu, C. Zhao, X. Qiao, C. Yang, et al., SOX17 loss-of-function mutation underlying familial pulmonary arterial hypertension, Int. Heart. J., 62 (2021), 566–574. https://doi.org/10.1536/ihj.20-711 doi: 10.1536/ihj.20-711
|
| [17] |
N. Zhu, C. L. Welch, J. Wang, P. M. Allen, C. Gonzaga-Jauregui, L. Ma, et al., Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease, Genome Med., 56 (2018). https://doi.org/10.1186/s13073-018-0566-x doi: 10.1186/s13073-018-0566-x
|
| [18] |
X. Yuan, Z. Wang, L. Wang, Q. Zhao, S. Gong, Y. Sun, et al., Increased levels of runt-related transcription factor 2 are associated with poor survival of patients with idiopathic pulmonary arterial hypertension, Am. J. Men's Health., 14 (2020). https://doi.org/10.1177/1557988320945458 doi: 10.1177/1557988320945458
|
| [19] |
L. C. Price, S. J. Wort, F. Perros, P. Dorfmüller, A. Huertas, D. Montani, et al., Inflammation in pulmonary arterial hypertension, Chest, 141 (2012), 210–221. https://doi.org/10.1378/chest.11-0793 doi: 10.1378/chest.11-0793
|
| [20] |
H. Zeng, X. Liu, Y. Zhang, Identification of potential biomarkers and immune infiltration characteristics in idiopathic pulmonary arterial hypertension using bioinformatics analysis, Front. Cardiovasc. Med., 8 (2021). https://doi.org/10.3389/fcvm.2021.624714 doi: 10.3389/fcvm.2021.624714
|
| [21] |
I. Sarrion, L. Milian, G. Juan, M. Ramon, I. Furest, C. Carda, et al., Role of circulating miRNAs as biomarkers in idiopathic pulmonary arterial hypertension: Possible relevance of miR-23a, Oxid. Med. Cell. Longevity, 2015 (2015), 792846. https://doi.org/10.1155/2015/792846 doi: 10.1155/2015/792846
|
| [22] |
W. He, X. Su, L. Chen, C. Liu, W. Lu, T. Wang, et al., Potential biomarkers and therapeutic targets of idiopathic pulmonary arterial hypertension, Physiol. Rep., 10 (2022), e15101. https://doi.org/10.14814/phy2.15101 doi: 10.14814/phy2.15101
|
| [23] |
C. Li, Z. Zhang, Q. Xu, R. Shi, Comprehensive analyses of miRNA-mRNA network and potential drugs in idiopathic pulmonary arterial hypertension, BioMed Res. Int., 2020 (2020), 5156304. https://doi.org/10.1155/2020/5156304 doi: 10.1155/2020/5156304
|
| [24] |
S. Hao, P. Jiang, L. Xie, G. Xiang, Z. Liu, W. Hu, et al., Essential genes and MiRNA-mRNA network contributing to the pathogenesis of idiopathic pulmonary arterial hypertension, Front. Cardiovasc. Med., 8 (2021), 627873. https://doi.org/10.3389/fcvm.2021.627873 doi: 10.3389/fcvm.2021.627873
|
| [25] |
D. Li, A. Tulahong, M. N. Uddin, H. Zhao, H. Zhang, Meta-analysis identifying epithelial-derived transcriptomes predicts poor clinical outcome and immune infiltrations in ovarian cancer, Math. Biosci. Eng., 18 (2021), 6527–6551. https://doi.org/10.3934/mbe.2021324 doi: 10.3934/mbe.2021324
|
| [26] |
E. Hsu, H. Shi, R. M. Jordan, J. Lyons-Weiler, J. M. Pilewski, C. A. Feghali-Bostwick, Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension, Arthritis Rheum., 63 (2011), 783–794. https://doi.org/10.1002/art.30159 doi: 10.1002/art.30159
|
| [27] |
L. Renaud, W. A. da Silveira, N. Takamura, G. Hardiman, C. Feghali-Bostwick, Prominence of IL6, IGF, TLR, and bioenergetics pathway perturbation in lung tissues of scleroderma patients with pulmonary fibrosis, front. immunol., 11 (2020), 383. https://doi.org/10.3389/fimmu.2020.00383 doi: 10.3389/fimmu.2020.00383
|
| [28] |
M. Mura, M. J. Cecchini, M. Joseph, J. T. Granton, Osteopontin lung gene expression is a marker of disease severity in pulmonary arterial hypertension, Respirology, 24 (2019), 1104–1110. https://doi.org/10.1111/resp.13557 doi: 10.1111/resp.13557
|
| [29] |
R. S. Stearman, Q. M. Bui, G. Speyer, A. Handen, A. R. Cornelius, B. B. Graham, et al., Systems analysis of the human pulmonary arterial hypertension lung transcriptome, Am. J. Respir. Cell Mol. Biol., 60 (2019), 637–649. https://doi.org/10.1165/rcmb.2018-0368OC doi: 10.1165/rcmb.2018-0368OC
|
| [30] |
C. E. Romanoski, X. Qi, S. Sangam, R. R. Vanderpool, R.S. Stearman, A. Conklin, et al., Transcriptomic profiles in pulmonary arterial hypertension associate with disease severity and identify novel candidate genes, Pulm. Circ., 10 (2020). https://doi.org/10.1177/2045894020968531 doi: 10.1177/2045894020968531
|
| [31] |
C. Cheadle, A. E. Berger, S. C. Mathai, D. N. Grigoryev, T. N. Watkins, Y. Sugawara, et al., Erythroid-specific transcriptional changes in PBMCs from pulmonary hypertension patients, PloS One., 7 (2012), e34951. https://doi.org/10.1371/journal.pone.0034951 doi: 10.1371/journal.pone.0034951
|
| [32] |
D. Wu, C. C. Talbot, Q. Liu, Z. Jing, R. L. Damico, R. Tuder et al., Identifying microRNAs targeting Wnt/β-catenin pathway in end-stage idiopathic pulmonary arterial hypertension, J. Mol. Med., 94 (2016), 875–885. https://doi.org/10.1007/s00109-016-1426-z doi: 10.1007/s00109-016-1426-z
|
| [33] |
J. Xia, E. E. Gill, R. E. W. Hancock, NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data, Nat. Protoc., 10 (2015), 823–844. https://doi.org/10.1038/nprot.2015.052 doi: 10.1038/nprot.2015.052
|
| [34] |
W. E. Johnson, C. Li, A. Rabinovic, Adjusting batch effects in microarray expression data using empirical bayes methods, Biostatistics, 8 (2007), 118–127. https://doi.org/10.1093/biostatistics/kxj037 doi: 10.1093/biostatistics/kxj037
|
| [35] |
M. E. Ritchie, B. Phipson, D. Wu, Y. Hu, C. W. Law, W. Shi, et al., limma powers differential expression analyses for RNA-sequencing and microarray studies, Nucleic Acids Res., 43 (2015), e47. https://doi.org/10.1093/nar/gkv007 doi: 10.1093/nar/gkv007
|
| [36] |
A. Subramanian, P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, et al., Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles, PNAS, 102 (2005), 15545–15550. https://doi.org/10.1073/pnas.0506580102 doi: 10.1073/pnas.0506580102
|
| [37] |
D. Szklarczyk, A. L. Gable, D. Lyon, A. Junge, S. Wyder, J. Huerta-Cepas, et al., STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets, Nucleic Acids Res., 47 (2019), 607–613. https://doi.org/10.1093/nar/gky1131 doi: 10.1093/nar/gky1131
|
| [38] |
C. Chin, S. Chen, H. Wu, C. Ho, M. Ko, C. Lin, CytoHubba: identifying hub objects and sub-networks from complex interactome, BMC Syst. Biol., 8 (2014), S11. https://doi.org/10.1186/1752-0509-8-S4-S11 doi: 10.1186/1752-0509-8-S4-S11
|
| [39] |
J. Wang, R. Akter, M. F. Shahriar, M. N. Uddin, Cancer-Associated Stromal Fibroblast-Derived Transcriptomes Predict Poor Clinical Outcomes and Immunosuppression in Colon Cancer, Pathol. Oncol. Res., (2022). https://doi.org/10.3389/pore.2022.1610350 doi: 10.3389/pore.2022.1610350
|
| [40] |
P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, et al., Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Res., 13 (2003), 2498–2504. https://doi.org/10.1101/gr.1239303 doi: 10.1101/gr.1239303
|
| [41] |
Y. Fan, K. Siklenka, S. K. Arora, P. Ribeiro, S. Kimmins, J. Xia, MiRNet-dissecting miRNA-target interactions and functional associations through network-based visual analysis, Nucleic Acids Res., 44 (2016), 135–141. https://doi.org/10.1093/nar/gkw288 doi: 10.1093/nar/gkw288
|
| [42] |
X. Robin, N. Turck, A. Hainard, N. Tiberti, F. Lisacek, J. Sanchez, M. Müller, PROC: an open-source package for R and S+ to analyze and compare ROC curves, BMC Bioinformatics., 77 (2011). https://doi.org/10.1186/1471-2105-12-77 doi: 10.1186/1471-2105-12-77
|
| [43] |
J. Wang, M. N. Uddin, J. Hao, R. Chen, Y. Xiang, D. Xiong, et al., Identification of potential novel prognosis-related genes through transcriptome sequencing, bioinformatics analysis, and clinical validation in acute myeloid leukemia, Front. Genet., 12 (2021). https://doi.org/10.3389/fgene.2021.723001 doi: 10.3389/fgene.2021.723001
|
| [44] |
M. N. Uddin, R. Akter, M. Li, Z. Abdelrahman, Expression of SARS-COV-2 cell receptor gene ACE2 is associated with immunosuppression and metabolic reprogramming in lung adenocarcinoma based on bioinformatics analyses of gene expression profiles, Chem. Biol. Interact., 335 (2021), 109370. https://doi.org/10.1016/j.cbi.2021.109370 doi: 10.1016/j.cbi.2021.109370
|
| [45] |
K. C. Cotto, A. H. Wagner, Y. Feng, S. Kiwala, A. C. Coffman, G. Spies, et al., DGIdb 3.0: A redesign and expansion of the drug-gene interaction database, Nucleic Acids Res., 46 (2018), 1068–1073. https://doi.org/10.1093/nar/gkx1143 doi: 10.1093/nar/gkx1143
|
| [46] |
X. Mao, Z. Ren, G. N. Parker, H. Sondermann, M. A. Pastorello, W. Wang, et al., Structural bases of unphosphorylated STAT1 association and receptor binding, Mol. Cell, 17 (2005), 761–771. https://doi.org/10.1016/j.molcel.2005.02.021 doi: 10.1016/j.molcel.2005.02.021
|
| [47] |
A. Yamamura, M. J. Nayeem, A. A. Mamun, R. Takahashi, H. Hayashi, M. Sato, Platelet-derived growth factor up-regulates Ca2+-sensing receptors in idiopathic pulmonary arterial hypertension, FASEB J., 33 (2019), 7363–7374. https://doi.org/10.1096/fj.201802620R doi: 10.1096/fj.201802620R
|
| [48] |
S. Gairhe, K. S. Awad, E. J. Dougherty, G. A. Ferreyra, S. Wang, Z. Yu, et al., Type Ⅰ interferon activation and endothelial dysfunction in caveolin-1 insufficiency-associated pulmonary arterial hypertension, PNAS, 118 (2021). https://doi.org/10.1073/pnas.2010206118 doi: 10.1073/pnas.2010206118
|
| [49] |
A. D. Stefano, G. Caramori, A. Capelli, I. Gnemmi, F. L. Ricciardolo, T. Oates, et al., STAT4 activation in smokers and patients with chronic obstructive pulmonary disease, Eur. Respir. J., 24 (2004), 78–85. https://doi.org/10.1183/09031936.04.00080303 doi: 10.1183/09031936.04.00080303
|
| [50] |
H. Alam, N. Li, S. S. Dhar, S. J. Wu, J. Lv, K. Chen, et al., HP1γ promotes lung adenocarcinoma by downregulating the transcription-repressive regulators NCOR2 and ZBTB7A, Cancer Res., 78 (2018), 3834–3848. https://doi.org/10.1158/0008-5472.CAN-17-3571 doi: 10.1158/0008-5472.CAN-17-3571
|
| [51] |
Y. Yang, H. Yuan, J. G. Edwards, Y. Skayian, K. Ochani, E. J. Miller, et al., Deletion of STAT5a/b in vascular smooth muscle abrogates the male bias in hypoxic pulmonary hypertension in mice: Implications in the human disease, Mol. Med., 20 (2014), 625–638. https://doi.org/10.2119/molmed.2014.00180 doi: 10.2119/molmed.2014.00180
|
| [52] |
T. Hashimoto-Kataoka, N. Hosen, T. Sonobe, Y. Arita, T. Yasui, T. Masaki, et al., Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension, PNAS, 112 (2015), 2677–2686. https://doi.org/10.1073/pnas.1424774112 doi: 10.1073/pnas.1424774112
|
| [53] |
E. Zhao, H. Xie, Y. Zhang, Identification of differentially expressed genes associated with idiopathic pulmonary arterial hypertension by integrated bioinformatics approaches, J. Comput. Biol., 28 (2021), 79–88. https://doi.org/10.1089/cmb.2019.0433 doi: 10.1089/cmb.2019.0433
|
| [54] |
W. Wang, Z. Jiang, D. Zhang, L. Fu, R. Wan, K. Hong, Comparative transcriptional analysis of pulmonary arterial hypertension associated with three different diseases, Front. Cell Dev. Biol., 9 (2021). https://doi.org/10.3389/fcell.2021.672159 doi: 10.3389/fcell.2021.672159
|
| [55] |
H. Göös, M. Kinnunen, K. Salokas, Z. Tan, X. Liu, L. Yadav, et al., Human transcription factor protein interaction networks, Nat. Commun., 13 (2022), 766. https://doi.org/10.1038/s41467-022-28341-5 doi: 10.1038/s41467-022-28341-5
|
| [56] |
Q. Yang, C. Jia, P. Wang, M. Xiong, J. Cui, L. Li, et al., MicroRNA-505 identified from patients with essential hypertension impairs endothelial cell migration and tube formation, Int. J. Cardiol., 177 (2014), 925–934. https://doi.org/10.1016/j.ijcard.2014.09.204 doi: 10.1016/j.ijcard.2014.09.204
|
| [57] |
H. Wang, Z. Ma, X. Liu, C. Zhang, Y. Hu, L. Ding, et al., MiR-183-5p is required for non-small cell lung cancer progression by repressing PTEN, Biomed. Pharmacother., 111 (2019), 1103–1111. https://doi.org/10.1016/j.biopha.2018.12.115 doi: 10.1016/j.biopha.2018.12.115
|
| [58] |
J. Li, S. Sun, N. Li, P. Lv, S. Xie, P. Wang, MiR-205 as a promising biomarker in the diagnosis and prognosis of lung cancer, Oncotarget, 8 (2017), 91938–91949. https://doi.org/10.18632/oncotarget.20262 doi: 10.18632/oncotarget.20262
|
| [59] |
Y. Zhao, J. Zhang, J. Yang, Y. Wei, J. Peng, C. Fu, et al., MiR-205-5p promotes lung cancer progression and is valuable for the diagnosis of lung cancer, Thorac Cancer, 13 (2022), 832–843. https://doi.org/10.1111/1759-7714.14331 doi: 10.1111/1759-7714.14331
|
| [60] |
W. Liu, X. Wan, Z. Mu, F. Li, L. Wang, J. Zhao, et al., MiR-1256 suppresses proliferation and migration of non-small cell lung cancer via regulating TCTN1, Oncol. Lett., 16 (2018), 1708–1714. https://doi.org/10.3892/ol.2018.8794 doi: 10.3892/ol.2018.8794
|
| [61] |
H. El Chami, P. M. Hassoun, Immune and inflammatory mechanisms in pulmonary arterial hypertension, Prog. Cardiovasc. Dis., 55 (2012), 218–228. https://doi.org/10.1016/j.pcad.2012.07.006 doi: 10.1016/j.pcad.2012.07.006
|
| [62] |
N. M. Patel, S. M. Kawut, S. Jelic, S. M. Arcasoy, D. J. Lederer, A. C. Borczuk, Pulmonary arteriole gene expression signature in idiopathic pulmonary fibrosis, Eur. Respir. J., 41 (2013), 1324–1330. https://doi.org/10.1183/09031936.00084112 doi: 10.1183/09031936.00084112
|
| [63] |
H. Yang, Y. Lu, H. Yang, Y. Zhu, Y. Tang, L. Li, et al., Integrated weighted gene co-expression network analysis uncovers STAT1(signal transducer and activator of transcription 1) and IFI44L (interferon-induced protein 44-like) as key genes in pulmonary arterial hypertension, Bioengineered, 12 (2021), 6021–6034. https://doi.org/10.1080/21655979.2021.1972200 doi: 10.1080/21655979.2021.1972200
|
| [64] |
L. Gabryšová, M. Alvarez-Martinez, R. Luisier, L. S. Cox, J. Sodenkamp, C. Hosking, et al., C-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4+ T cells, Nat. Immunol., 19 (2018), 497–507. https://doi.org/10.1038/s41590-018-0083-5 doi: 10.1038/s41590-018-0083-5
|
| [65] |
X. Yang, C. Wang, Y. Lin, P. Zhang, Identification of crucial hub genes and differential T cell infiltration in idiopathic pulmonary arterial hypertension using bioinformatics strategies, Front. Mol. Biosci., 9 (2022). https://doi.org/10.3389/fmolb.2022.800888 doi: 10.3389/fmolb.2022.800888
|
| [66] |
S. Ni, T. Ji, J. Dong, F. Chen, H. Feng, H. Zhao, et al., Immune cells in pulmonary arterial hypertension, Heart Lung Circ., 31 (2022), 934–943. https://doi.org/10.1016/j.hlc.2022.02.007 doi: 10.1016/j.hlc.2022.02.007
|
| [67] |
M. Rabinovitch, C. Guignabert, M. Humbert, M. R. Nicolls, Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension, Circ. Res., 115 (2014), 165–175. https://doi.org/10.1161/CIRCRESAHA.113.301141 doi: 10.1161/CIRCRESAHA.113.301141
|
| [68] |
M. Masullo, M. Menegazzi, S. Di Micco, P. Beffy, G. Bifulco, M. Dal Bosco, et al., Direct interaction of garcinol and related polyisoprenylated benzophenones of Garcinia cambogia fruits with the transcription factor STAT-1 as a likely mechanism of their inhibitory effect on cytokine signaling pathways, J. Nat. Prod., 77 (2014), 543–549. https://doi.org/10.1021/np400804y doi: 10.1021/np400804y
|
| [69] |
M. Toshner, E. Spiekerkoetter, H. Bogaard, G. Hansmann, S. Nikkho, K. W. Prins, Repurposing of medications for pulmonary arterial hypertension, Pulm. Circ., 10 (2020). https://doi.org/10.1177/2045894020941494 doi: 10.1177/2045894020941494
|
| [70] |
R. Papp, C. Nagaraj, D. Zabini, B. M. Nagy, M. Lengyel, D. S. Maurer, et al., Targeting TMEM16A to reverse vasoconstriction and remodelling in idiopathic pulmonary arterial hypertension, Eur. Respir. J., 53 (2019). https://doi.org/10.1183/13993003.00965-2018 doi: 10.1183/13993003.00965-2018
|
 mbe-20-02-194 supplementary.xlsx mbe-20-02-194 supplementary.xlsx |
 |








Figures(9) / Tables(3)

Qian Li, Minawaer Hujiaaihemaiti, Jie Wang, Md. Nazim Uddin, Ming-Yuan Li, Alidan Aierken, Yun Wu. Identifying key transcription factors and miRNAs coregulatory networks associated with immune infiltrations and drug interactions in idiopathic pulmonary arterial hypertension[J]. Mathematical Biosciences and Engineering, 2023, 20(2): 4153-4177. doi: 10.3934/mbe.2023194







 DownLoad:
DownLoad: