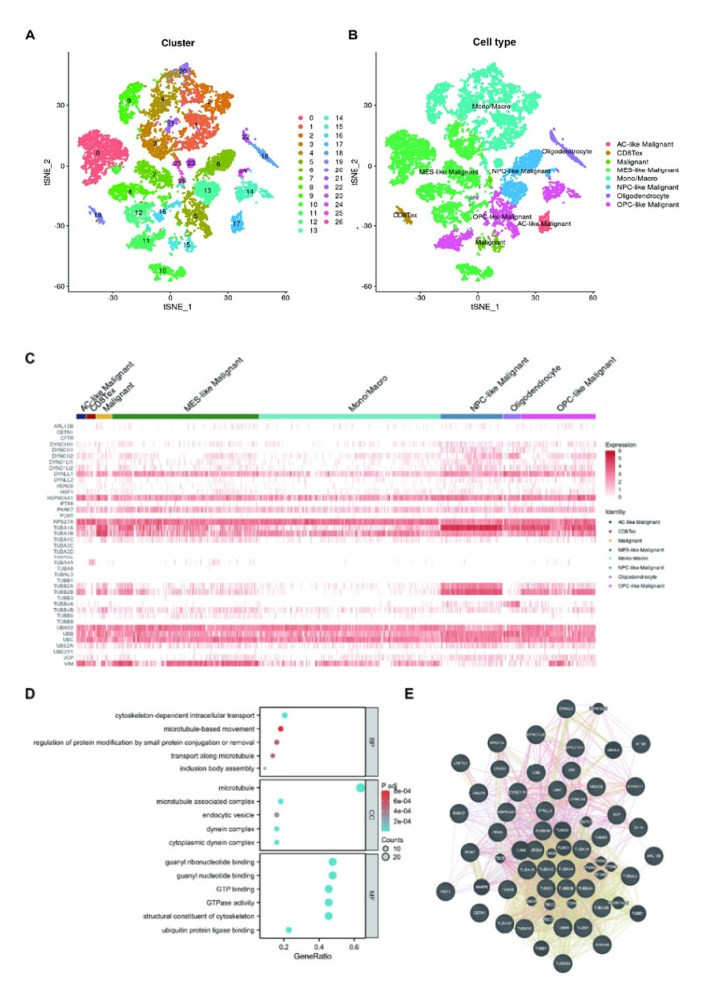
Background: Aggrephagy is a lysosome-dependent process that degrades misfolded protein condensates to maintain cancer cell homeostasis. Despite its importance in cellular protein quality control, the role of aggrephagy in glioma remains poorly understood. Objective: To investigate the expression of aggrephagy-related genes (ARGs) in glioma and in different cell types of gliomas and to develop an ARGs-based prognostic signature to predict the prognosis, tumor microenvironment, and immunotherapy response of gliomas. Methods: ARGs were identified by searching the Reactome database. We developed the ARGs-based prognostic signature (ARPS) using data from the Cancer Genome Atlas (TCGA, n = 669) by Lasso-Cox regression. We validated the robustness of the signature in clinical subgroups and CGGA cohorts (n = 970). Gene set enrichment analysis (GSEA) was used to identify the pathways enriched in ARPS subgroups. The correlations between ARGs and macrophages were also investigated at single cell level. Results: A total of 44 ARGs showed heterogeneous expression among different cell types of gliomas. Five ARGs (HSF1, DYNC1H1, DYNLL2, TUBB6, TUBA1C) were identified to develop ARPS, an independent prognostic factor. GSEA showed gene sets of patients with high-ARPS were mostly enriched in cell cycle, DNA replication, and immune-related pathways. High-ARPS subgroup had higher immune cell infiltration states, particularly macrophages, Treg cells, and neutrophils. APRS had positive association with tumor mutation burden (TMB) and immunotherapy response predictors. At the single cell level, we found ARGs correlated with macrophage development and identified ARGs-mediated macrophage subtypes with distinct communication characteristics with tumor cells. VIM+ macrophages were identified as pro-inflammatory and had higher interactions with malignant cells. Conclusion: We identified a novel signature based on ARGs for predicting glioma prognosis, tumor microenvironment, and immunotherapy response. We highlight the ARGs-mediated macrophages in glioma exhibit classical features.
Citation: Xiaowei Zhang, Jiayu Tan, Xinyu Zhang, Kritika Pandey, Yuqing Zhong, Guitao Wu, Kejun He. Aggrephagy-related gene signature correlates with survival and tumor-associated macrophages in glioma: Insights from single-cell and bulk RNA sequencing[J]. Mathematical Biosciences and Engineering, 2024, 21(2): 2407-2431. doi: 10.3934/mbe.2024106
Background: Aggrephagy is a lysosome-dependent process that degrades misfolded protein condensates to maintain cancer cell homeostasis. Despite its importance in cellular protein quality control, the role of aggrephagy in glioma remains poorly understood. Objective: To investigate the expression of aggrephagy-related genes (ARGs) in glioma and in different cell types of gliomas and to develop an ARGs-based prognostic signature to predict the prognosis, tumor microenvironment, and immunotherapy response of gliomas. Methods: ARGs were identified by searching the Reactome database. We developed the ARGs-based prognostic signature (ARPS) using data from the Cancer Genome Atlas (TCGA, n = 669) by Lasso-Cox regression. We validated the robustness of the signature in clinical subgroups and CGGA cohorts (n = 970). Gene set enrichment analysis (GSEA) was used to identify the pathways enriched in ARPS subgroups. The correlations between ARGs and macrophages were also investigated at single cell level. Results: A total of 44 ARGs showed heterogeneous expression among different cell types of gliomas. Five ARGs (HSF1, DYNC1H1, DYNLL2, TUBB6, TUBA1C) were identified to develop ARPS, an independent prognostic factor. GSEA showed gene sets of patients with high-ARPS were mostly enriched in cell cycle, DNA replication, and immune-related pathways. High-ARPS subgroup had higher immune cell infiltration states, particularly macrophages, Treg cells, and neutrophils. APRS had positive association with tumor mutation burden (TMB) and immunotherapy response predictors. At the single cell level, we found ARGs correlated with macrophage development and identified ARGs-mediated macrophage subtypes with distinct communication characteristics with tumor cells. VIM+ macrophages were identified as pro-inflammatory and had higher interactions with malignant cells. Conclusion: We identified a novel signature based on ARGs for predicting glioma prognosis, tumor microenvironment, and immunotherapy response. We highlight the ARGs-mediated macrophages in glioma exhibit classical features.

| [1] |
A. Darlix, S. Zouaoui, V. Rigau, F. Bessaoud, D. Figarella-Branger, H. Mathieu-Daudé, et al., Epidemiology for primary brain tumors: a nationwide population-based study, J. Neuro-Oncol., 131 (2017), 525–546. https://doi.org/10.1007/s11060-016-2318-3 doi: 10.1007/s11060-016-2318-3

|
| [2] |
Q. T. Ostrom, L. Bauchet, F. G. Davis, I. Deltour, J. L. Fisher, C. E. Langer, et al., The epidemiology of glioma in adults: a "state of the science" review, Neuro-Oncology, 16 (2014), 896–913. https://doi.org/10.1093/neuonc/nou087 doi: 10.1093/neuonc/nou087
|
| [3] |
R. Stupp, W. P. Mason, M. J. van den Bent, M. Welle, B. Fisher, B. Fisher, et al., Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma, N. Engl. J. Med., 352 (2005), 987–996. https://doi.org/10.1056/NEJMoa043330 doi: 10.1056/NEJMoa043330
|
| [4] |
J. M. Hyttinen, M. Amadio, J. Viiri, A. Pascale, A. Salminen, K. Kaarniranta, et al., Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases, Ageing Res. Rev., 18 (2014), 16–28. https://doi.org/10.1016/j.arr.2014.07.002 doi: 10.1016/j.arr.2014.07.002
|
| [5] |
J. S. Valastyan, S. Lindquist, Mechanisms of protein-folding diseases at a glance, Dis. Models Mech., 7 (2014), 9–14. https://doi.org/10.1242/dmm.013474 doi: 10.1242/dmm.013474
|
| [6] |
N. Gregersen, P. Bross, S. Vang, J. H. Christensen, Protein misfolding and human disease, Annu. Rev. Genomics Hum. Genet., 7 (2006), 103–124. https://doi.org/10.1146/annurev.genom.7.080505.115737 doi: 10.1146/annurev.genom.7.080505.115737
|
| [7] |
J. Vaquer-Alicea, M. I. Diamond, Propagation of protein aggregation in neurodegenerative diseases, Annu. Rev. Biochem., 88 (2019), 785–810. https://doi.org/10.1146/annurev-biochem-061516-045049 doi: 10.1146/annurev-biochem-061516-045049
|
| [8] |
A. V. Kumar, J. Mills, L. R. Lapierre, Selective autophagy receptor p62/SQSTM1, a pivotal player in stress and aging, Front. Cell Dev. Biol., 10 (2022), 793328. https://doi.org/10.3389/fcell.2022.793328 doi: 10.3389/fcell.2022.793328
|
| [9] |
T. Lamark, T. Johansen, Aggrephagy: selective disposal of protein aggregates by macroautophagy, Int. J. Cell Biol., 2012 (2012). https://doi.org/10.1155/2012/736905 doi: 10.1155/2012/736905
|
| [10] |
X. Ma, C. Lu, Y. Chen, S. Li, N. Ma, X. Tao, et al., CCT2 is an aggrephagy receptor for clearance of solid protein aggregates, Cell, 185 (2022), 1325–1345.e22. https://doi.org/10.1016/j.cell.2022.03.005 doi: 10.1016/j.cell.2022.03.005
|
| [11] |
M. H. Z. Guang, E. L. Kavanagh, L. P. Dunne, P. Dowling, L. Zhang, S. Lindsay, et al., Targeting proteotoxic stress in cancer: a review of the role that protein quality control pathways play in oncogenesis, Cancers (Basel), 11 (2019), 66. https://doi.org/10.3390/cancers11010066 doi: 10.3390/cancers11010066
|
| [12] |
A. Pataer, B. Ozpolat, R. Shao, N. R. Cashman, S. S. Plotkin, C. E. Samuel, et al., Therapeutic targeting of the PI4K2A/PKR lysosome network is critical for misfolded protein clearance and survival in cancer cells, Oncogene, 39 (2020), 801–813. https://doi.org/10.1038/s41388-019-1010-4 doi: 10.1038/s41388-019-1010-4
|
| [13] |
Y. C. Tsai, A. M. Weissman, The unfolded protein response, degradation from the endoplasmic reticulum, and cancer, Genes Cancer, 1 (2010), 764–778. https://doi.org/10.1177/1947601910383011 doi: 10.1177/1947601910383011
|
| [14] |
T. Simms-Waldrip, A. Rodriguez-Gonzalez, T. Lin, A. K. Ikeda, C. Fu, K. M. Sakamoto, The aggresome pathway as a target for therapy in hematologic malignancies, Mol. Genet. Metab., 94 (2008), 283–286. https://doi.org/10.1016/j.ymgme.2008.03.012 doi: 10.1016/j.ymgme.2008.03.012
|
| [15] |
S. T. Nawrocki, J. S. Carew, M. S. Pino, R. A. Highshaw, R. H. I. Andtbacka, K. Dunner Jr., et al., Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells, Cancer Res., 66 (2006), 3773–3781. https://doi.org/10.1158/0008-5472.CAN-05-2961 doi: 10.1158/0008-5472.CAN-05-2961
|
| [16] |
N. Amer, H. Taha, D. Hesham, N. Al-Shehaby, A. Mosaab, M. Soudy, et al., Aggresomes predict poor outcomes and implicate proteostasis in the pathogenesis of pediatric choroid plexus tumors, J. Neuro-Oncol., 152 (2021), 67–78. https://doi.org/10.1007/s11060-020-03694-3 doi: 10.1007/s11060-020-03694-3
|
| [17] |
L. Simone, F. Pisani, M. G. Mola, M. De Bellis, G. Merla, L. Micale, et al., AQP4 aggregation state is a determinant for glioma cell fate, Cancer Res., 79 (2019), 2182–2194. https://doi.org/10.1158/0008-5472.CAN-18-2015 doi: 10.1158/0008-5472.CAN-18-2015
|
| [18] |
D. Wang, Y. Jiang, T. Wang, Z. Wang, F. Zou, Identification of a novel autophagy-related prognostic signature and small molecule drugs for glioblastoma by bioinformatics, BMC Med. Genomics, 15 (2022), 111. https://doi.org/10.1186/s12920-022-01261-5 doi: 10.1186/s12920-022-01261-5
|
| [19] |
S. Feng, H. Liu, X. Dong, P. Du, H. Guo, Q. Pang, Identification and validation of an autophagy-related signature for predicting survival in lower-grade glioma, Bioengineered, 12 (2021), 9692–9708. https://doi.org/10.1080/21655979.2021.1985818 doi: 10.1080/21655979.2021.1985818
|
| [20] |
Y. Fan, X. Peng, B. Li, G. Zhao, Development of autophagy signature-based prognostic nomogram for refined glioma survival prognostication, BioMed Res. Int., 2020 (2020), 1872962. https://doi.org/10.1155/2020/1872962 doi: 10.1155/2020/1872962
|
| [21] |
T. Wu, E. Hu, S. Xu, M. Chen, P. Guo, Z. Dai, et al., clusterProfiler 4.0: A universal enrichment tool for interpreting omics data, Innovation, 2 (2021), 100141. https://doi.org/10.1016/j.xinn.2021.100141 doi: 10.1016/j.xinn.2021.100141
|
| [22] |
K. Yoshihara, M. Shahmoradgoli, E. Martínez, R. Vegesna, H. Kim, W. Torres-Garcia, et al., Inferring tumour purity and stromal and immune cell admixture from expression data, Nat. Commun., 4 (2013), 2612. https://doi.org/10.1038/ncomms3612 doi: 10.1038/ncomms3612
|
| [23] |
A. M. Newman, C. L. Liu, M. R. Green, A. J. Gentles, W. Feng, Y. Xu, et al., Robust enumeration of cell subsets from tissue expression profiles, Nat. Methods, 12 (2015), 453–457. https://doi.org/10.1038/nmeth.3337 doi: 10.1038/nmeth.3337
|
| [24] |
D. Zeng, Z. Ye, R. Shen, G. Yu, J. Wu, Y. Xiong, et al., IOBR: Multi-omics immuno-oncology biological research to decode tumor microenvironment and signatures, Front. Immunol., 12 (2021), 687975. https://doi.org/10.3389/fimmu.2021.687975 doi: 10.3389/fimmu.2021.687975
|
| [25] |
Ó. Lapuente-Santana, M. Van Genderen, P. A. J. Hilbers, F. Finotello, F. Eduati, Interpretable systems biomarkers predict response to immune-checkpoint inhibitors, Patterns, 2 (2021), 100293. https://doi.org/10.1016/j.patter.2021.100293 doi: 10.1016/j.patter.2021.100293
|
| [26] |
M. S. Rooney, S. A. Shukla, C. J. Wu, G. Getz, N. Hacohen, Molecular and genetic properties of tumors associated with local immune cytolytic activity, Cell, 160 (2015), 48–61. https://doi.org/10.1016/j.cell.2014.12.033 doi: 10.1016/j.cell.2014.12.033
|
| [27] |
R. Cabrita, M. Lauss, A. Sanna, M. Donia, M. S. Larsen, S. Mitra, et al., Tertiary lymphoid structures improve immunotherapy and survival in melanoma, Nature, 577 (2020), 561–565. https://doi.org/10.1038/s41586-019-1914-8 doi: 10.1038/s41586-019-1914-8
|
| [28] |
M. Ayers, J. Lunceford, M. Nebozhyn, E. Murphy, A. Loboda, D. R. Kaufman, et al., IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade, J. Clin. Invest., 127 (2017), 2930–2940. https://doi.org/10.1172/JCI91190 doi: 10.1172/JCI91190
|
| [29] |
W. Roh, P. L. Chen, A. Reuben, C. N. Spencer, P. A. Prieto, J. P. Miller, et al., Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance, Sci. Transl. Med., 9 (2017). https://doi.org/10.1126/scitranslmed.aah3560 doi: 10.1126/scitranslmed.aah3560
|
| [30] |
T. Davoli, H. Uno, E. C. Wooten, S. J. Elledge, Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy, Science, 355 (2017). https://doi.org/10.1126/science.aaf8399 doi: 10.1126/science.aaf8399
|
| [31] |
J. L. Messina, D. A. Fenstermacher, S. Eschrich, X. Qu, A. E. Berglund, M. C. Lloyd, et al., 12-Chemokine gene signature identifies lymph node-like structures in melanoma: Potential for patient selection for immunotherapy, Sci. Rep., 2 (2012), 765. https://doi.org/10.1038/srep00765 doi: 10.1038/srep00765
|
| [32] |
P. Jiang, S. Gu, D. Pan, J. Fu, A. Sahu, X. Hu, et al., Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response, Nat. Med., 24 (2018), 1550–1558. https://doi.org/10.1038/s41591-018-0136-1 doi: 10.1038/s41591-018-0136-1
|
| [33] |
C. Neftel, J. Laffy, M. G. Filbin, T. Hara, M. E. Shore, G. J. Rahme, et al., An integrative model of cellular states, plasticity, and genetics for glioblastoma, Cell, 178 (2019), 835–849.e21. https://doi.org/10.1016/j.cell.2019.06.024 doi: 10.1016/j.cell.2019.06.024
|
| [34] |
D. Sun, J. Wang, Y. Han, X. Dong, J. Ge, R. Zheng, et al., TISCH: a comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment, Nucleic Acids Res., 49 (2021), D1420–D1430. https://doi.org/10.1093/nar/gkaa1020 doi: 10.1093/nar/gkaa1020
|
| [35] |
X. Qiu, Q. Mao, Y. Tang, L. Wang, R. Chawla, H. A. Pliner, et al., Reversed graph embedding resolves complex single-cell trajectories, Nat. Methods, 14 (2017), 979–982. https://doi.org/10.1038/nmeth.4402 doi: 10.1038/nmeth.4402
|
| [36] |
Y. Chen, J. Yin, W. Li, H. Li, D. Chen, C. Zhang, et al., Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma, Cell Res., 30 (2020), 1024–1042. https://doi.org/10.1038/s41422-020-0374-x doi: 10.1038/s41422-020-0374-x
|
| [37] |
Y. Gao, H. Wang, S. Chen, R. An, Y. Chu, G. Li, et al., Single-cell N(6)-methyladenosine regulator patterns guide intercellular communication of tumor microenvironment that contribute to colorectal cancer progression and immunotherapy, J. Transl. Med., 20 (2022), 197. https://doi.org/10.1186/s12967-022-03395-7 doi: 10.1186/s12967-022-03395-7
|
| [38] |
S. Jin, C. F. Guerrero-Juarez, L. Zhang, I. Chang, R. Ramos, C. Kuan, et al., Inference and analysis of cell-cell communication using CellChat, Nat. Commun., 12 (2021), 1088. https://doi.org/10.1038/s41467-021-21246-9 doi: 10.1038/s41467-021-21246-9
|
| [39] |
Y. Wu, S. Yang, J. Ma, Z. Chen, G. Song, D. Rao, et al., Spatiotemporal immune landscape of colorectal cancer liver metastasis at single-cell level, Cancer Discovery, 12 (2022), 134–153. https://doi.org/10.1158/2159-8290.CD-21-0316 doi: 10.1158/2159-8290.CD-21-0316
|
| [40] |
Y. Watanabe, A. Tsujimura, K. Taguchi, M. Tanaka, HSF1 stress response pathway regulates autophagy receptor SQSTM1/p62-associated proteostasis, Autophagy, 13 (2017), 133–148. https://doi.org/10.1080/15548627.2016.1248018 doi: 10.1080/15548627.2016.1248018
|
| [41] |
G. Wang, P. Cao, Y. Fan, K. Tan, Emerging roles of HSF1 in cancer: Cellular and molecular episodes, Biochim. Biophys. Acta, Rev. Cancer, 1874 (2020), 188390. https://doi.org/10.1016/j.bbcan.2020.188390 doi: 10.1016/j.bbcan.2020.188390
|
| [42] |
B. Dai, A. Gong, Z. Jing, K. D. Aldape, S. Kang, R. Sawaya, et al., Forkhead box M1 is regulated by heat shock factor 1 and promotes glioma cells survival under heat shock stress, J. Biol. Chem., 288 (2013), 1634–1642. https://doi.org/10.1074/jbc.M112.379362 doi: 10.1074/jbc.M112.379362
|
| [43] |
P. Antonietti, B. Linder, S. Hehlgans, I. C. Mildenberger, M. C. Burger, S. Fulda, et al., Interference with the HSF1/HSP70/BAG3 pathway primes glioma cells to matrix detachment and BH3 mimetic–induced apoptosis, Mol. Cancer Ther., 16 (2017), 156–168. https://doi.org/10.1158/1535-7163.MCT-16-0262 doi: 10.1158/1535-7163.MCT-16-0262
|
| [44] |
H. T. Hoang, M. A. Schlager, A. P. Carter, S. L. Bullock, DYNC1H1 mutations associated with neurological diseases compromise processivity of dynein-dynactin-cargo adaptor complexes, PNAS, 114 (2017), E1597–E1606. https://doi.org/10.1073/pnas.1620141114 doi: 10.1073/pnas.1620141114
|
| [45] |
T. Furukawa, Y. Kuboki, E. Tanji, S. Yoshida, T. Hatori, M. Yamamoto, et al., Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas, Sci. Rep., 1 (2011), 161. https://doi.org/10.1038/srep00161 doi: 10.1038/srep00161
|
| [46] |
J. Bai, B. Yang, R. Shi, X. Shao, Y. Yang, F. Wang, et al., Could microtubule inhibitors be the best choice of therapy in gastric cancer with high immune activity: mutant DYNC1H1 as a biomarker, Aging, 12 (2020), 25101–25119. https://doi.org/10.18632/aging.104084 doi: 10.18632/aging.104084
|
| [47] |
J. Jiang, D. Liu, G. Xu, T. Liang, C. Yu, S. Liao, et al., TRIM68, PIKFYVE, and DYNLL2: The possible novel autophagy- and immunity-associated gene biomarkers for osteosarcoma prognosis, Front. Oncol., 11 (2021), 643104. https://doi.org/10.3389/fonc.2021.643104 doi: 10.3389/fonc.2021.643104
|
| [48] |
A. E. Gylfe, J. Kondelin, M. Turunen, H. Ristolainen, R. Katainen, E. Pitkänen, et al., Identification of candidate oncogenes in human colorectal cancers with microsatellite instability, Gastroenterology, 145 (2013), 540–543.E22. https://doi.org/10.1053/j.gastro.2013.05.015 doi: 10.1053/j.gastro.2013.05.015
|
| [49] |
J. Zhang, J. Y. Huang, Y. N. Chen, F. Yuan, H. Zhang, F. H. Yan, et al., Whole genome and transcriptome sequencing of matched primary and peritoneal metastatic gastric carcinoma, Sci. Rep., 5 (2015), 13750. https://doi.org/10.1038/srep15309 doi: 10.1038/srep15309
|
| [50] |
D. Zhang, J. Zhao, C. Han, X. Liu, J. Liu, H. Yang, et al., Identification of hub genes related to prognosis in glioma, Biosci. Rep., 40 (2020). https://doi.org/10.1042/BSR20193377 doi: 10.1042/BSR20193377
|
| [51] |
M. A. H. Albahde, P. Zhang, Q. Zhang, G. Li, W. Wang, Upregulated expression of TUBA1C predicts poor prognosis and promotes oncogenesis in pancreatic ductal adenocarcinoma via regulating the cell cycle, Front. Oncol., 10 (2020). https://doi.org/10.3389/fonc.2020.00049 doi: 10.3389/fonc.2020.00049
|
| [52] |
T. Bian, M. Zheng, D. Jiang, J. Liu, H. Sun, X. Li, et al., Prognostic biomarker TUBA1C is correlated to immune cell infiltration in the tumor microenvironment of lung adenocarcinoma, Cancer Cell Int., 21 (2021), 144. https://doi.org/10.1186/s12935-021-01849-4 doi: 10.1186/s12935-021-01849-4
|
| [53] |
C. C. N. Wang, C. Y. Li, J. H. Cai, P. C. Y. Sheu, J. J. P. Tsai, M. Y. Meng, et al., Identification of prognostic candidate genes in breast cancer by integrated bioinformatic analysis, J. Clin. Med., 8 (2019). https://doi.org/10.3390/jcm8081160 doi: 10.3390/jcm8081160
|
| [54] |
Y. Li, Q. Liang, Y. Q. Wen, L. L. Chen, L. T. Wang, Y. L. Liu, et al., Comparative proteomics analysis of human osteosarcomas and benign tumor of bone, Cancer Genet. Cytogenet., 198 (2010), 97–106. https://doi.org/10.1016/j.cancergencyto.2010.01.003 doi: 10.1016/j.cancergencyto.2010.01.003
|
| [55] |
H. Zhu, X. Hu, L. Gu, Z. Jian, L. Li, S. Hu, et al., TUBA1C is a prognostic marker in low-grade glioma and correlates with immune cell infiltration in the tumor microenvironment, Front. Genet., 12 (2021), 759953. https://doi.org/10.3389/fgene.2021.759953 doi: 10.3389/fgene.2021.759953
|
| [56] |
W. H. Fridman, L. Zitvogel, C. Sautès-Fridman, G. Kroemer, The immune contexture in cancer prognosis and treatment, Nat. Rev. Clin. Oncol., 14 (2017), 717–734. https://doi.org/10.1038/nrclinonc.2017.101 doi: 10.1038/nrclinonc.2017.101
|
| [57] |
Z. Duan, Y. Luo, Targeting macrophages in cancer immunotherapy, Signal Transduction Targeted Ther., 6 (2021), 127. https://doi.org/10.1038/s41392-021-00506-6 doi: 10.1038/s41392-021-00506-6
|
| [58] |
D. H. Josephs, H. J. Bax, S. N. Karagiannis, Tumour-associated macrophage polarisation and re-education with immunotherapy, Front. Biosci. Elite, 7 (2015), 293–308. https://doi.org/10.2741/e735 doi: 10.2741/e735
|
| [59] |
J. R. Conway, E. Kofman, S. S. Mo, H. Elmarakeby, E. Van Allen, Genomics of response to immune checkpoint therapies for cancer: implications for precision medicine, Genome Med., 10 (2018), 1–18. https://doi.org/10.1186/s13073-018-0605-7 doi: 10.1186/s13073-018-0605-7
|
| [60] |
Q. Jia, H. Chu, Z. Jin, H. Long, B. Zhu, High-throughput single-сell sequencing in cancer research, Signal Transduction Targeted Ther., 7 (2022), 145. https://doi.org/10.1038/s41392-022-00990-4 doi: 10.1038/s41392-022-00990-4
|
| [61] |
P. Li, X. Kong, Y. He, Y. Liu, X. Peng, Z. Li, et al., Recent developments in application of single-cell RNA sequencing in the tumour immune microenvironment and cancer therapy, Mil. Med. Res., 9 (2022), 52. https://doi.org/10.1186/s40779-022-00414-y doi: 10.1186/s40779-022-00414-y
|
| [62] |
X. Zhang, Single-cell RNA sequencing identifies macrophage signatures correlated with clinical features and tumour microenvironment in meningiomas, IET Syst. Biol., 17 (2023), 259–270. https://doi.org/10.1049/syb2.12074 doi: 10.1049/syb2.12074
|
| [63] |
L. Håversen, J. P. Sundelin, A. Mardinoglu, M. Rutberg, M. Ståhlman, U. Wilhelmsson, et al., Vimentin deficiency in macrophages induces increased oxidative stress and vascular inflammation but attenuates atherosclerosis in mice, Sci. Rep., 8 (2018), 16973. https://doi.org/10.1038/s41598-018-34659-2 doi: 10.1038/s41598-018-34659-2
|
| [64] |
N. Mor-Vaknin, A. Punturieri, K. Sitwala, D. M. Markovitz, Vimentin is secreted by activated macrophages, Nat. Cell Biol., 5 (2003), 59–63. https://doi.org/10.1038/ncb898 doi: 10.1038/ncb898
|
 mbe-21-02-106-supplementary.pdf mbe-21-02-106-supplementary.pdf |
 |







Figures(8) / Tables(1)

Xiaowei Zhang, Jiayu Tan, Xinyu Zhang, Kritika Pandey, Yuqing Zhong, Guitao Wu, Kejun He. Aggrephagy-related gene signature correlates with survival and tumor-associated macrophages in glioma: Insights from single-cell and bulk RNA sequencing[J]. Mathematical Biosciences and Engineering, 2024, 21(2): 2407-2431. doi: 10.3934/mbe.2024106







 DownLoad:
DownLoad: