The tumor immune microenvironment of colorectal cancer (CRC) affects tumor development, prognosis and immunotherapy strategies. Recently, immune-related lncRNA were shown to play vital roles in the tumor immune microenvironment. The objective of this study was to identify lncRNAs involved in the immune response, tumorigenesis and progression of CRC and to establish an immune-related lncRNA signature for predicting the prognosis of CRC.
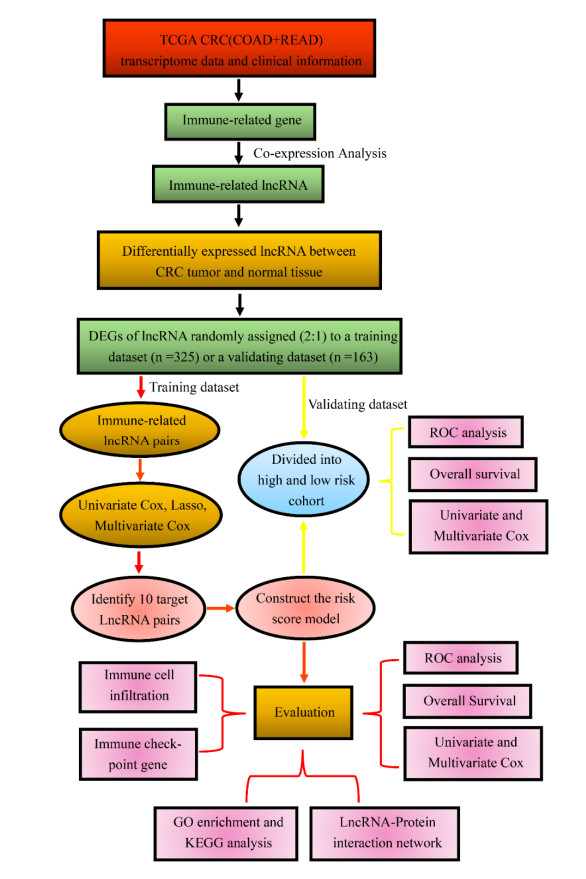
We used data retrieved from the cancer genome atlas (TCGA) dataset to construct a 10-gene immune-related lncRNA pair (IRLP) signature model using a method based on the ranking and comparison of paired gene expression in CRC. The clinical prognosis, immune checkpoints and lncRNA-protein networks were analyzed to evaluate the signature.
The signature was closely associated with overall survival of CRC patients (p < 0.001 in both of the training and validating cohorts) and the 3-year AUC values for the training and validating cohorts were 0.884 and 0.739, respectively. And, there were positive correlations between the signature and age (p = 0.048), clinical stage (p < 0.01), T stage (p < 0.01), N stage (p < 0.001) and M stage (p < 0.01). In addition, the signature model appeared to be highly relevant to some checkpoints, including CD160, TNFSF15, HHLA2, IDO2 and KIR3DL1. Further, molecular functional analysis and lncRNA-protein networks were applied to understand the molecular mechanisms underlying the carcinogenic effect and progression.
The 10-gene IRLP signature model is an independent prognostic factor for CRC patient and can be utilized for the development of immunotherapy.
Citation: Bin Ma, Lianqun Cao, Yongmin Li. A novel 10-gene immune-related lncRNA signature model for the prognosis of colorectal cancer[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 9743-9760. doi: 10.3934/mbe.2021477
The tumor immune microenvironment of colorectal cancer (CRC) affects tumor development, prognosis and immunotherapy strategies. Recently, immune-related lncRNA were shown to play vital roles in the tumor immune microenvironment. The objective of this study was to identify lncRNAs involved in the immune response, tumorigenesis and progression of CRC and to establish an immune-related lncRNA signature for predicting the prognosis of CRC.
We used data retrieved from the cancer genome atlas (TCGA) dataset to construct a 10-gene immune-related lncRNA pair (IRLP) signature model using a method based on the ranking and comparison of paired gene expression in CRC. The clinical prognosis, immune checkpoints and lncRNA-protein networks were analyzed to evaluate the signature.
The signature was closely associated with overall survival of CRC patients (p < 0.001 in both of the training and validating cohorts) and the 3-year AUC values for the training and validating cohorts were 0.884 and 0.739, respectively. And, there were positive correlations between the signature and age (p = 0.048), clinical stage (p < 0.01), T stage (p < 0.01), N stage (p < 0.001) and M stage (p < 0.01). In addition, the signature model appeared to be highly relevant to some checkpoints, including CD160, TNFSF15, HHLA2, IDO2 and KIR3DL1. Further, molecular functional analysis and lncRNA-protein networks were applied to understand the molecular mechanisms underlying the carcinogenic effect and progression.
The 10-gene IRLP signature model is an independent prognostic factor for CRC patient and can be utilized for the development of immunotherapy.

| [1] |
F. Bray, J. Ferlay, I. Soerjomataram, R. Siegel, L. Torre, A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J. Clin., 68 (2018), 394-424. doi: 10.3322/caac.21492

|
| [2] | A. Jemal, E. M. Ward, C. J. Johnson, K. A. Cronin, J. Ma, B. Ryerson, et al., Annual report to the nation on the status of cancer, 1975-2014, featuring survival, J. Natl. Cancer Inst., 109 (2017). |
| [3] |
L. Bertero, F. Massa, J. Metovic, R. Zanetti, I. Castellano, U. Ricardi, et al., Eighth Edition of the UICC Classification of Malignant Tumours: an overview of the changes in the pathological TNM classification criteria-What has changed and why?, Virchows Arch., 472 (2018), 519-531. doi: 10.1007/s00428-017-2276-y
|
| [4] |
S. Perakis, J. Thomas, M. Pichler, Non-coding RNAs enabling prognostic stratification and prediction of therapeutic response in colorectal cancer patients, Adv. Exp. Med. Biol., 937 (2016), 183-204. doi: 10.1007/978-3-319-42059-2_10
|
| [5] |
Y. Liu, R. Jing, J. Xu, K. Liu, J. Xue, Z. Wen, et al., Comparative analysis of oncogenes identified by microarray and RNA-sequencing as biomarkers for clinical prognosis, Biomark Med., 9 (2015), 1067-1078. doi: 10.2217/bmm.15.97
|
| [6] |
R. Spizzo, M. I. Almeida, A. Colombatti, G. A. Calin, Long non-coding RNAs and cancer: a new frontier of translational research?, Oncogene, 31(2012), 4577-4587. doi: 10.1038/onc.2011.621
|
| [7] |
D. Dimartino, A. Colantoni, M. Ballarino, J. Martone, D. Mariani, J. Danner, et al., The long non-coding RNA lnc-31 interacts with rock1 mRNA and mediates its YB-1-dependent translation, Cell Rep., 23 (2018), 733-740. doi: 10.1016/j.celrep.2018.03.101
|
| [8] |
S. H. E. Kaufmann, Immunology's Coming of Age, Front Immunol., 10 (2019), 684. doi: 10.3389/fimmu.2019.00684
|
| [9] |
Y. Chen, A. Satpathy, H. Chang, Gene regulation in the immune system by long noncoding RNAs, Nat. Immunol., 18 (2017), 962-972. doi: 10.1038/ni.3771
|
| [10] |
J. Xu, Q. Meng, X. Li, H. Yang, J. Xu, N. Gao, et al., Long noncoding RNA mir17HG promotes colorectal cancer progression via miR-17-5p, Cancer Res., 79 (2019), 4882-4895. doi: 10.1158/0008-5472.CAN-18-3880
|
| [11] |
S. Djebali, C. Davis, A. Merkel, A. Dobin, T. Lassmann, A. Mortazavi, et al., Landscape of transcription in human cells, Nature, 489 (2012), 101-108. doi: 10.1038/nature11233
|
| [12] |
A. M. Newman, C. L. Liu, M. R. Green, A. J. Gentles, W. Feng, Y. Xu, et al., Robust enumeration of cell subsets from tissue expression profiles, Nat. Methods, 12 (2015), 453-457. doi: 10.1038/nmeth.3337
|
| [13] | R. Dienstmann, L. Vermeulen, J. Guinney, S. Kopetz, S. Tejpar, J. Tabernero, Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer, 17 (2017), 268. |
| [14] |
X. Wang, Q. Jin, X. Wang, W. Chen, Z. Cai, LncRNA ZFAS1 promotes proliferation and migration and inhibits apoptosis in nasopharyngeal carcinoma via the PI3K/AKT pathway in vitro, Cancer Biomark., 26 (2019), 171-182.15. doi: 10.3233/CBM-182080
|
| [15] |
X. Jia, P. Niu, C. Xie, H. Liu, Long noncoding RNA PXN-AS1-L promotes the malignancy of nasopharyngeal carcinoma cells via upregulation of SAPCD2, Cancer Med., 8 (2019), 4278-4291. doi: 10.1002/cam4.2227
|
| [16] |
H. Kambara, F. Niazi, L. Kostadinova, D. K. Moonka, C. T. Siegel, A. B. Post, et al., Negative regulation of the interferon response by an interferon-induced long non-coding RNA, Nucleic. Acids Res., 42 (2014), 10668-10680. doi: 10.1093/nar/gku713
|
| [17] |
P. Wang, Y. Xue, Y. Han, L. Lin, C. Wu, S. Xu, et al., The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation, Science, 344 (2014), 310-313. doi: 10.1126/science.1251456
|
| [18] |
G. Hu, Q. Tang, Su. Sharma, F. Yu, T. M. Escobar, S. A. Muljo, et al., Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation, Nat. Immunol., 14 (2013), 1190-1198. doi: 10.1038/ni.2712
|
| [19] |
Q. Chen, C. Wei, Z. X. Wang, M. Sun, Long non-coding RNAs in anti-cancer drug resistance, Oncotarget, 8 (2017), 1925-1936. doi: 10.18632/oncotarget.12461
|
| [20] | M. Mu, Y. Tang, Z. Yang, Y. Qiu, X. Li, W. Mo, et al., Effect of different expression of immune-related lncRNA on colon adenocarcinoma and its relation to prognosis, Biomed. Res. Int., 2020 (2020), 6942740. |
| [21] |
Y. Wang, J. Liu, F. Ren, Y. Chu, B. Cui, Identification and validation of a four-long non-coding RNA signature associated with immune infiltration and prognosis in colon cancer, Front. Genet., 12 (2021), 671128. doi: 10.3389/fgene.2021.671128
|
| [22] |
F. Qin, H. Xu, G. Wei, Y. Ji, J. Yu, C. Hu, et al., A prognostic model based on the immune-related lncRNAs in colorectal cancer, Front. Genet., 12 (2021), 658736. doi: 10.3389/fgene.2021.658736
|
| [23] |
M. Sun, T. Zhang, Y. Wang, W. Huang, L. Xia, A novel signature constructed by immune-related lncRNA predicts the immune landscape of colorectal cancer, Front. Genet., 12 (2021), 695130. doi: 10.3389/fgene.2021.695130
|
| [24] |
J. A. Eddy, J. Sung, D. Geman, N. D. Price, Relative expression analysis for molecular cancer diagnosis and prognosis, Technol. Cancer Res. Treat., 9 (2010), 149-159. doi: 10.1177/153303461000900204
|
| [25] |
J. Luo, P. Liu, L. Wang, Y. Huang, Y. Wang, W. Geng, et al., Establishment of an immune-related gene pair model to predict colon adenocarcinoma prognosis, BMC Cancer, 20 (2020), 1071. doi: 10.1186/s12885-020-07532-7
|
| [26] |
X. Mo, X. Huang, Y. Feng, C. Wei, H. Liu, H. Ru, et al., Immune infiltration and immune gene signature predict the response to fluoropyrimidine-based chemotherapy in colorectal cancer patients, Oncoimmunology, 9 (2020), 1832347. doi: 10.1080/2162402X.2020.1832347
|
| [27] | S. P. Arlauckas, C. S. Garris, R. H. Kohler, M. Kitaoka, M. F. Cuccarese, K. S. Yang, et al., In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy, Sci. Transl. Med., 9 (2017), |
| [28] |
A. Furukawa, M. Meguro, R. Yamazaki, H. Watanabe, A. Takahashi, K. Kuroki, et al., Evaluation of the reactivity and receptor competition of HLA-G isoforms toward available antibodies: implications of structural characteristics of hla-g isoforms, Int. J. Mol. Sci., 20 (2019), 5947. doi: 10.3390/ijms20235947
|
| [29] |
Y. Jiang, O. Chen, C. Cui, B. Zhao, X. Han, Z. Zhang, et al., KIR3DS1/L1 and HLA-Bw4-80I are associated with HIV disease progression among HIV typical progressors and long-term nonprogressors, BMC Infect. Dis., 13 (2013), 405. doi: 10.1186/1471-2334-13-405
|
| [30] |
H. Sun, J. Xu, Q. Huang, M. Huang, K. Li, K. Qu, et al., Correction: reduced CD160 expression contributes to impaired NK-cell function and poor clinical outcomes in patients with HCC, Cancer Res., 79 (2019), 1714. doi: 10.1158/0008-5472.CAN-19-0630
|
| [31] |
Xu G, Shi Y, Ling X, Wang D, Liu Y, Lu H, et al., HHLA2 predicts better survival and exhibits inhibited proliferation in epithelial ovarian cancer, Cancer Cell Int., 21 (2021), 252. doi: 10.1186/s12935-021-01930-y
|
| [32] |
Y. Liu, P. Xu, H. Liu, C. Fang, H. Guo, X. Chen, et al., Silencing IDO2 in dendritic cells: A novel strategy to strengthen cancer immunotherapy in a murine lung cancer model, Int. J. Oncol., 57 (2020), 587-597. doi: 10.3892/ijo.2020.5073
|
| [33] |
J. Bayry, Immunology: TL1A in the inflammatory network in autoimmune diseases, Nat. Rev. Rheumatol., 6 (2010), 67-68. doi: 10.1038/nrrheum.2009.263
|
| [34] |
W. Hou, D. Medynski, S. Wu, X. Lin, L. Y. Li, VEGI-192, a new isoform of TNFSF15, specifically eliminates tumor vascular endothelial cells and suppresses tumor growth, Clin. Cancer Res., 11 (2005), 5595-5602. doi: 10.1158/1078-0432.CCR-05-0384
|
| [35] |
C. Cavallini, O. Lovato, A. Bertolaso, E. Zoratti, G. Malpeli, E. Mimiola, et al., Expression and function of the TL1A/DR3 axis in chronic lymphocytic leukemia, Oncotarget, 6 (2015), 32061-32074. doi: 10.18632/oncotarget.5201
|
| [36] |
N. Zhang, P. Wu, D. Shayiremu, L. Wu, H. Shan, L. Ye, et al., Suppression of renal cell carcinoma growth in vivo by forced expression of vascular endothelial growth inhibitor, Int. J. Oncol., 42 (2013), 1664-1673. doi: 10.3892/ijo.2013.1877
|
| [37] |
W. Dong, Z. Cao, Y. Pang, T. Feng, H. Tian, CARF, as an oncogene, promotes colorectal cancer stemness by activating erbb signaling pathway, Onco. Targets Ther., 12 (2019), 9041-9051. doi: 10.2147/OTT.S225733
|
| [38] |
L. Barault, N. Veyrie, V. Jooste, D. Lecorre, C. Chapusot, J. Ferraz, et al., Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers, Int. J. Cancer, 122 (2008), 2255-2259. doi: 10.1002/ijc.23388
|
| [39] |
Ogino S, Nosho K, Kirkner GJ, Shima K, Irahara N, Kure S, et al., PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer, J Clin. Oncol, 27 (2009), 1477-1484. doi: 10.1200/JCO.2008.18.6544
|
| [40] |
M. H. Hofmann, M. Gmachl, J. Ramharter, F. Savarese, D. Gerlach, J. R. Marszalek, et al., BI-3406, a Potent and Selective SOS1-KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition, Cancer Discov., 11 (2021), 142-157. doi: 10.1158/2159-8290.CD-20-0142
|
| [41] |
N. Ishaque, M. L. Abba, C. Hauser, N. Patil, N. Paramasivam, D. Huebschmann, et al., Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer, Nat. Commun., 9 (2018), 4782. doi: 10.1038/s41467-018-07041-z
|
| [42] |
S. Cascio, O. J. Finn, Complex of MUC1, CIN85 and Cbl in colon cancer progression and metastasis, Cancers (Basel), 7 (2015), 342-352. doi: 10.3390/cancers7010342
|
| [43] |
W. Dai, Y. Xu, S. Mo, Q. Li, J. Yu, R. Wang, et al., GLUT3 induced by AMPK/CREB1 axis is key for withstanding energy stress and augments the efficacy of current colorectal cancer therapies, Signal Transduct Tar. Ther., 5 (2020), 177. doi: 10.1038/s41392-020-00220-9
|
| [44] |
C. Zhuo, D. Hu, J. Li, H. Yu, X. Lin, Y. Chen, et al., Downregulation of activin a receptor type 2a is associated with metastatic potential and poor prognosis of colon cancer, J. Cancer, 9 (2018), 3626-3633. doi: 10.7150/jca.26790
|
| [45] | R. Zhou, Y. Huang, B. Cheng, Y. Wang, B. Xiong, TGFBR1*6A is a potential modifier of migration and invasion in colorectal cancer cells, Oncol. Lett., 15 (2018), 3971-3976. |
| [46] |
T. S. Freedman, H. Sondermann, G. D. Friedland, T. Kortemme, D. Bar-Sagi, S. Marqusee, et al., A Ras-induced conformational switch in the Ras activator Son of sevenless, Proc. Natl. Acad. Sci. USA, 103 (2006), 16692-16697. doi: 10.1073/pnas.0608127103
|
| [47] |
H. Jeng, L. J. Taylor, D. Bar-Sagi, Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis, Nat. Commun., 3 (2012), 1168. doi: 10.1038/ncomms2173
|







Figures(8) / Tables(1)

Bin Ma, Lianqun Cao, Yongmin Li. A novel 10-gene immune-related lncRNA signature model for the prognosis of colorectal cancer[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 9743-9760. doi: 10.3934/mbe.2021477







 DownLoad:
DownLoad: