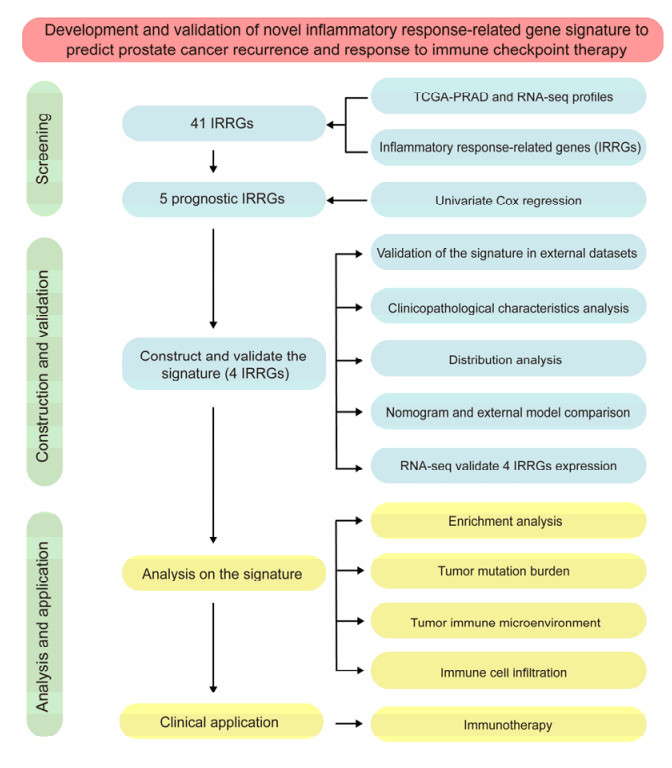
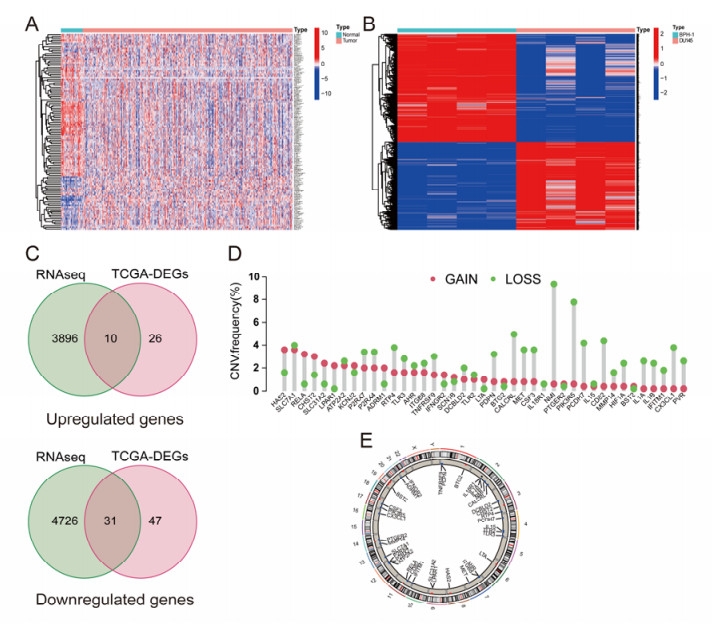
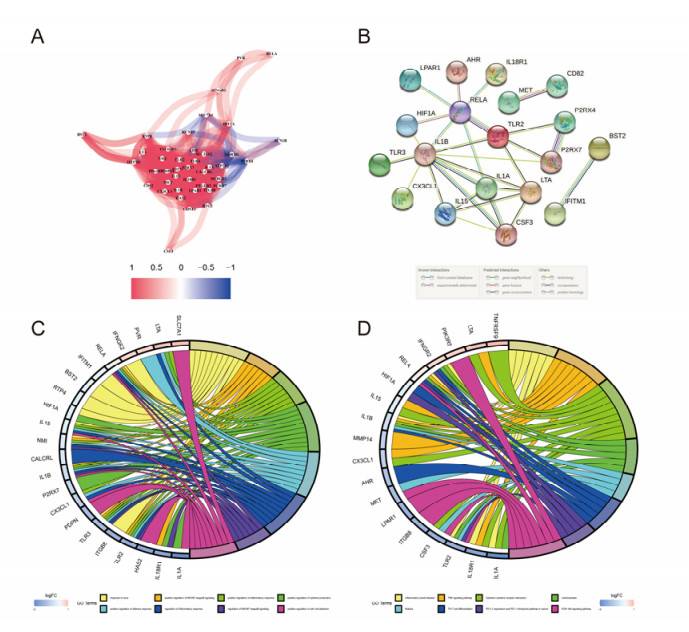
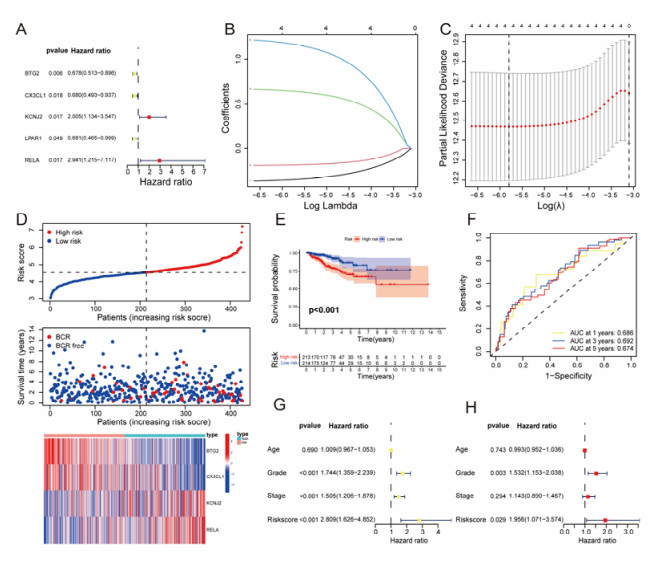
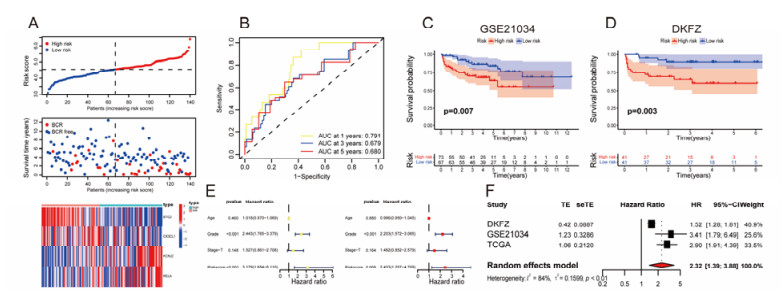
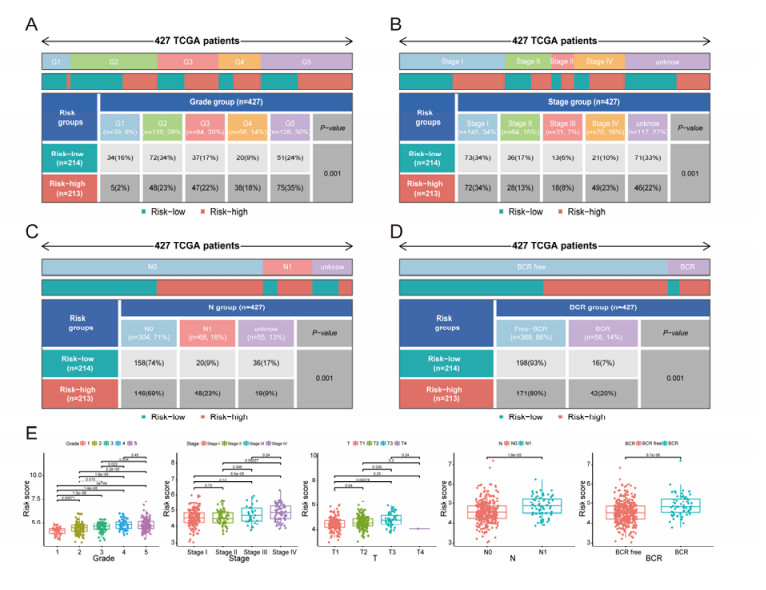
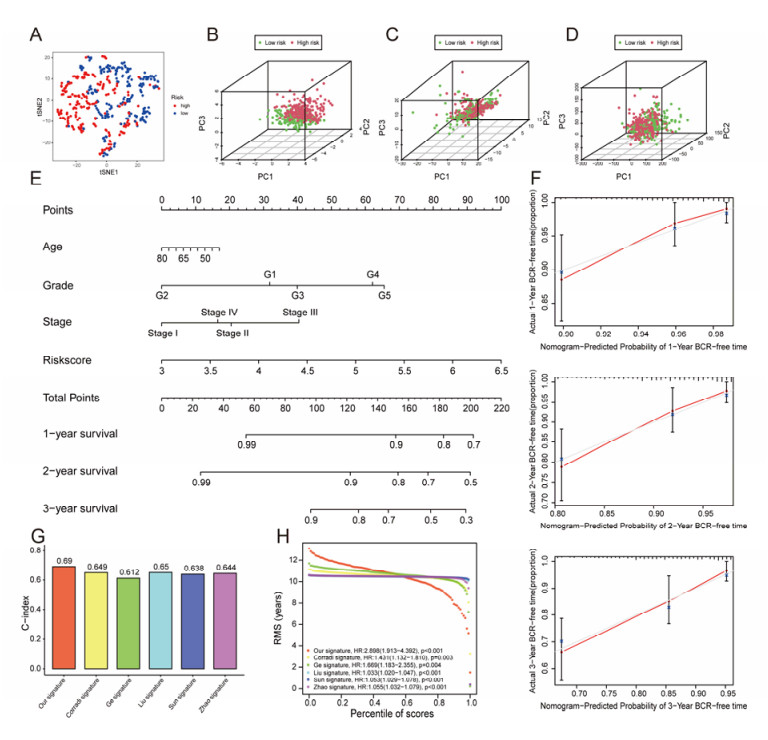
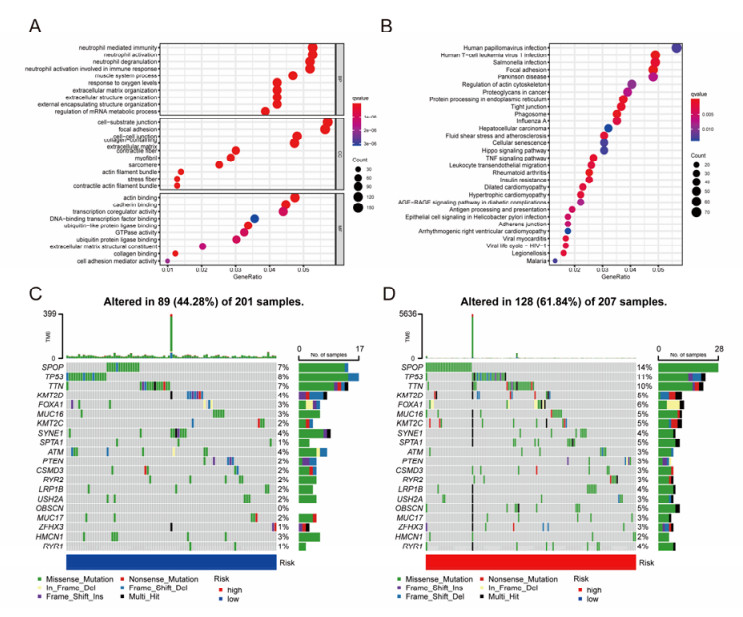
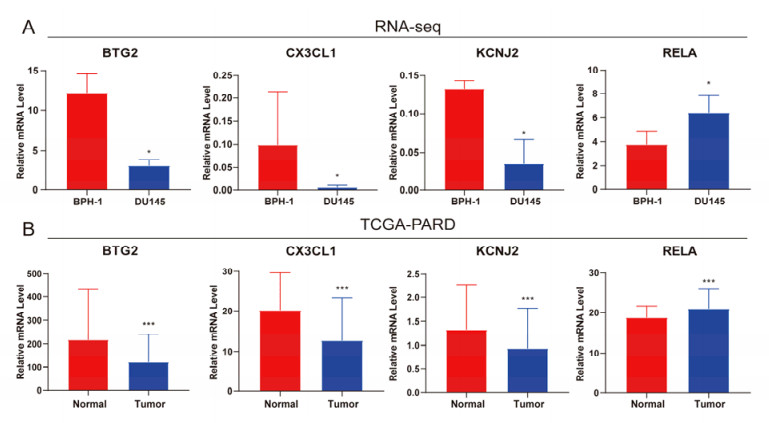
The aim of this study is to construct an inflammatory response-related genes (IRRGs) signature to monitor biochemical recurrence (BCR) and treatment effects in prostate cancer patients (PCa). A gene signature for inflammatory responses was constructed on the basis of the data from the Cancer Genome Atlas (TCGA) database, and validated in external datasets. It was analyzed using receiver operating characteristic curve, BCR-free survival, Cox regression, and nomogram. Distribution analysis and external model comparison were utilized. Then, enrichment analysis, tumor mutation burden, tumor immune microenvironment, and immune cell infiltration signatures were investigated. The role of the signature in immunotherapy was evaluated. The expression patterns of core genes were verified by RNA sequencing. We identified an IRRGs signature in the TCGA-PRAD cohort and verified it well in two other independent external datasets. The signature was a robust and independent prognostic index for predicting the BCR of PCa. The high-risk group of our signature predicted a shortened BCR time and an aggressive disease progression. A nomogram was constructed to predict BCR-free time in clinical practices. Neutrophils and CD8+ T cells were in higher abundance among the low-risk individuals. Immune functions varied significantly between the two groups and immune checkpoint therapy worked better for the low-risk patients. The expression of four IRRGs showed significant differences between PCa and surrounding benign tissues, and were validated in BPH-1 and DU145 cell lines by RNA sequencing. Our signature served as a reliable and promising biomarker for predicting the prognosis and evaluating the efficacy of immunotherapy, facilitating a better outcome for PCa patients.
Citation: Yong Luo, Xiaopeng Liu, Jingbo Lin, Weide Zhong, Qingbiao Chen. Development and validation of novel inflammatory response-related gene signature to predict prostate cancer recurrence and response to immune checkpoint therapy[J]. Mathematical Biosciences and Engineering, 2022, 19(11): 11345-11366. doi: 10.3934/mbe.2022528
The aim of this study is to construct an inflammatory response-related genes (IRRGs) signature to monitor biochemical recurrence (BCR) and treatment effects in prostate cancer patients (PCa). A gene signature for inflammatory responses was constructed on the basis of the data from the Cancer Genome Atlas (TCGA) database, and validated in external datasets. It was analyzed using receiver operating characteristic curve, BCR-free survival, Cox regression, and nomogram. Distribution analysis and external model comparison were utilized. Then, enrichment analysis, tumor mutation burden, tumor immune microenvironment, and immune cell infiltration signatures were investigated. The role of the signature in immunotherapy was evaluated. The expression patterns of core genes were verified by RNA sequencing. We identified an IRRGs signature in the TCGA-PRAD cohort and verified it well in two other independent external datasets. The signature was a robust and independent prognostic index for predicting the BCR of PCa. The high-risk group of our signature predicted a shortened BCR time and an aggressive disease progression. A nomogram was constructed to predict BCR-free time in clinical practices. Neutrophils and CD8+ T cells were in higher abundance among the low-risk individuals. Immune functions varied significantly between the two groups and immune checkpoint therapy worked better for the low-risk patients. The expression of four IRRGs showed significant differences between PCa and surrounding benign tissues, and were validated in BPH-1 and DU145 cell lines by RNA sequencing. Our signature served as a reliable and promising biomarker for predicting the prognosis and evaluating the efficacy of immunotherapy, facilitating a better outcome for PCa patients.

| [1] |
H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal, et al., Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J. Clin., 71 (2021), 209–249. https://doi.org/10.3322/caac.21660 doi: 10.3322/caac.21660

|
| [2] |
C. Xia, X. Dong, H. Li, M. Cao, D. Sun, S. He, et al., Cancer statistics in China and United States, 2022: Profiles, trends, and determinants, Chin. Med. J. Peking, 135 (2022), 584–590. https://doi.org/10.1097/CM9.0000000000002108 doi: 10.1097/CM9.0000000000002108
|
| [3] |
R. L. Siegel, K. D. Miller, H. E. Fuchs, A. Jemal, Cancer statistics, 2022, CA Cancer J. Clin., 72 (2022), 7–33. https://doi.org/10.3322/caac.21708 doi: 10.3322/caac.21708
|
| [4] |
S. R. Hawken, D. E. Spratt, J. Qi, S. M. Linsell, M. L. Cher, K. R. Ghani, et al., Utilization of salvage radiation therapy for biochemical recurrence after radical prostatectomy, Int. J. Radiat. Oncol. Biol. Phys., 104 (2019), 1030–1034. https://doi.org/10.1016/j.ijrobp.2019.01.006 doi: 10.1016/j.ijrobp.2019.01.006
|
| [5] |
J. D. Wolchok, H. Kluger, M. K. Callahan, M. A. Postow, N. A. Rizvi, A. M. Lesokhin, et al., Nivolumab plus ipilimumab in advanced melanoma, N. Engl. J. Med., 369 (2013), 122–133. https://doi.org/10.1056/NEJMoa1302369 doi: 10.1056/NEJMoa1302369
|
| [6] |
P. M. Ellis, E. T. Vella, Y. C. Ung, Immune checkpoint inhibitors for patients with advanced non-small-cell lung cancer: A systematic review, Clin. Lung Cancer, 18 (2017), 444–459. https://doi.org/10.1016/j.cllc.2017.02.001 doi: 10.1016/j.cllc.2017.02.001
|
| [7] |
M. Rouanne, M. Roumiguié, N. Houédé, A. Masson-Lecomte, P. Colin, G. Pignot, et al., Development of immunotherapy in bladder cancer: Present and future on targeting PD(L)1 and CTLA-4 pathways, World J. Urol., 36 (2018), 1727–1740. https://doi.org/10.1007/s00345-018-2332-5 doi: 10.1007/s00345-018-2332-5
|
| [8] |
S. Venkatachalam, T. R. McFarland, N. Agarwal, U. Swami, Immune checkpoint inhibitors in prostate cancer, Cancers, 13 (2021), 2187. https://doi.org/10.3390/cancers13092187 doi: 10.3390/cancers13092187
|
| [9] |
F. Massari, C. Ciccarese, A. Caliò, E. Munari, L. Cima, A. B. Porcaro, et al., Magnitude of PD-1, PD-L1 and T lymphocyte expression on tissue from castration-resistant prostate adenocarcinoma: An exploratory analysis, Targeted Oncol, 11 (2016), 345–351. https://doi.org/10.1007/s11523-015-0396-3 doi: 10.1007/s11523-015-0396-3
|
| [10] |
W. Baas, S. Gershburg, D. Dynda, K. Delfino, K. Robinson, D. Nie, et al., Immune characterization of the programmed death receptor pathway in high risk prostate cancer, Clin. Genitourin. Cancer, 15 (2017), 577–581. https://doi.org/10.1016/j.clgc.2017.04.002 doi: 10.1016/j.clgc.2017.04.002
|
| [11] |
R. M. Zemek, W. L. Chin, A. K. Nowak, M. J. Millward, R. A. Lake, W. J. Lesterhuis, Sensitizing the tumor microenvironment to immune checkpoint therapy, Front. Immunol., 11 (2020), 223. https://doi.org/10.3389/fimmu.2020.00223 doi: 10.3389/fimmu.2020.00223
|
| [12] |
K. D. Runcie, M. C. Dallos, Prostate cancer immunotherapy-finally in from the cold? Curr. Oncol. Rep., 23 (2021), 88. https://doi.org/10.1007/s11912-021-01084-0 doi: 10.1007/s11912-021-01084-0
|
| [13] |
H. Cha, J. H. Lee, S. Ponnazhagan, Revisiting immunotherapy: A focus on prostate cancer, Cancer Res., 80 (2020), 1615–1623. https://doi.org/10.1158/0008-5472.CAN-19-2948 doi: 10.1158/0008-5472.CAN-19-2948
|
| [14] |
M. Binnewies, E. W. Roberts, K. Kersten, V. Chan, D. F. Fearon, M. Merad, et al., Understanding the tumor immune microenvironment (TIME) for effective therapy, Nat. Med., 24 (2018), 541–550. https://doi.org/10.1038/s41591-018-0014-x doi: 10.1038/s41591-018-0014-x
|
| [15] |
K. S. Sfanos, S. Yegnasubramanian, W. G. Nelson, A. M. De Marzo, The inflammatory microenvironment and microbiome in prostate cancer development, Nat. Rev. Urol., 15 (2018), 11–24. https://doi.org/10.1038/nrurol.2017.167 doi: 10.1038/nrurol.2017.167
|
| [16] |
M. Archer, N. Dogra, N. Kyprianou, Inflammation as a driver of prostate cancer metastasis and therapeutic resistance, Cancers, 12 (2020), 2984. https://doi.org/10.3390/cancers12102984 doi: 10.3390/cancers12102984
|
| [17] |
L. Ferrero-Miliani, O. H. Nielsen, P. S. Andersen, S. E. Girardin, Chronic inflammation: Importance of NOD2 and NALP3 in interleukin-1beta generation, Clin. Exp. Immunol., 147 (2007), 227–235. https://doi.org/10.1111/j.1365-2249.2006.03261.x doi: 10.1111/j.1365-2249.2006.03261.x
|
| [18] |
A. K. Tewari, J. A. Stockert, S. S. Yadav, K. K. Yadav, I. Khan, Inflammation and prostate cancer, Adv. Exp. Med. Biol., 1095 (2018), 41–65. https://doi.org/10.1007/978-3-319-95693-0_3 doi: 10.1007/978-3-319-95693-0_3
|
| [19] |
A. Mayakonda, D. Lin, Y. Assenov, C. Plass, H. P. Koeffler, Maftools: Efficient and comprehensive analysis of somatic variants in cancer, Genome Res., 28 (2018), 1747–1756. https://doi.org/10.1101/gr.239244.118 doi: 10.1101/gr.239244.118
|
| [20] |
H. Zhang, P. Meltzer, S. Davis, RCircos: An R package for circos 2D track plots, BMC Bioinf., 14 (2013), 244. https://doi.org/10.1186/1471-2105-14-244 doi: 10.1186/1471-2105-14-244
|
| [21] |
M. E. Ritchie, B. Phipson, D. Wu, Y. Hu, C. W. Law, W. Shi, et al., Limma powers differential expression analyses for RNA-sequencing and microarray studies, Nucleic Acids Res., 43 (2015), e47. https://doi.org/10.1093/nar/gkv007 doi: 10.1093/nar/gkv007
|
| [22] |
Y. Luo, G. Zhang, Identification of a necroptosis-related prognostic index and associated regulatory axis in kidney renal clear cell carcinoma, Int. J. Gen. Med., 15 (2022), 5407–5423. https://doi.org/10.2147/IJGM.S367173 doi: 10.2147/IJGM.S367173
|
| [23] | Gene Ontology Consortium, Gene Ontology Consortium: Going forward, Nucleic Acids Res., 43 (2015), D1049–D1056. https://doi.org/10.1093/nar/gku1179 |
| [24] |
G. Yu, L. Wang, Y. Han, Q. He, ClusterProfiler: An R package for comparing biological themes among gene clusters, Omics J. Integr. Biol., 16 (2012), 284–287. https://doi.org/10.1089/omi.2011.0118 doi: 10.1089/omi.2011.0118
|
| [25] |
M. Kanehisa, S. Goto, KEGG: Kyoto encyclopedia of genes and genomes, Nucleic Acids Res., 28 (2000), 27–30. https://doi.org/10.1093/nar/28.1.27 doi: 10.1093/nar/28.1.27
|
| [26] |
T. Li, J. Fan, B. Wang, N. Traugh, Q. Chen, J. S. Liu, et al., TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells, Cancer Res., 77 (2017), e108–e110. https://doi.org/10.1158/0008-5472.CAN-17-0307 doi: 10.1158/0008-5472.CAN-17-0307
|
| [27] |
Y. Luo, Q. Chen, J. Lin, Identification and validation of a tumor mutation burden-related signature combined with immune microenvironment infiltration in adrenocortical carcinoma, Math. Biosci. Eng., 19 (2022), 7055–7075. https://doi.org/10.3934/mbe.2022333 doi: 10.3934/mbe.2022333
|
| [28] |
A. Grosset, V. Ouellet, C. Caron, G. Fragoso, V. Barrès, N. Delvoye, et al., Validation of the prognostic value of NF-κB P65 in prostate cancer: A retrospective study using a large multi-institutional cohort of the Canadian prostate cancer biomarker network, PLoS Med., 16 (2019), e1002847. https://doi.org/10.1371/journal.pmed.1002847 doi: 10.1371/journal.pmed.1002847
|
| [29] |
H. Xu, Q. Ding, H. Jiang, Genetic Polymorphism of interleukin-1A (IL-1A), IL-1B, and IL-1 receptor antagonist (IL-1RN) and prostate cancer risk, Asian Pac. J. Cancer Prev., 15 (2014), 8741–8747. https://doi.org/10.7314/apjcp.2014.15.20.8741 doi: 10.7314/apjcp.2014.15.20.8741
|
| [30] |
J. P. Corradi, C. W. Cumarasamy, I. Staff, J. Tortora, A. Salner, T. McLaughlin, et al., Identification of a five gene signature to predict time to biochemical recurrence after radical prostatectomy, Prostate, 81 (2021), 694–702. https://doi.org/10.1002/pros.24150 doi: 10.1002/pros.24150
|
| [31] |
S. Ge, X. Hua, J. Chen, H. Xiao, L. Zhang, J. Zhou, et al., Identification of a costimulatory molecule-related signature for predicting prognostic risk in prostate cancer, Front. Genet., 12 (2021), 666300. https://doi.org/10.3389/fgene.2021.666300 doi: 10.3389/fgene.2021.666300
|
| [32] |
H. Liu, L. Gao, T. Xie, J. Li, T. Zhai, Y. Xu, Identification and validation of a prognostic signature for prostate cancer based on ferroptosis-related genes, Front. Oncol., 11 (2021), 623313. https://doi.org/10.3389/fonc.2021.623313 doi: 10.3389/fonc.2021.623313
|
| [33] |
Z. Sun, Y. Mao, X. Zhang, S. Lu, H. Wang, C. Zhang, et al., Identification of ARHGEF38, NETO2, GOLM1, and SAPCD2 associated with prostate cancer progression by bioinformatic analysis and experimental validation, Front. Cell Dev. Biol., 9 (2021), 718638. https://doi.org/10.3389/fcell.2021.718638 doi: 10.3389/fcell.2021.718638
|
| [34] |
H. Zhao, Y. Zeng, Z. Han, Y. Zhuo, Y. Liang, C. T. Hon, et al., Novel immune-related signature for risk stratification and prognosis in prostatic adenocarcinoma, Cancer Sci., 112 (2021), 4365–4376. https://doi.org/10.1111/cas.15062 doi: 10.1111/cas.15062
|
| [35] |
S. E. Jalava, A. Urbanucci, L. Latonen, K. K. Waltering, B. Sahu, O. A. Jänne, et al., Androgen-regulated miR-32 targets BTG2 and is overexpressed in castration-resistant prostate cancer, Oncogene, 31 (2012), 4460–4471. https://doi.org/10.1038/onc.2011.624 doi: 10.1038/onc.2011.624
|
| [36] |
P. Liu, Y. Liang, L. Jiang, H. Wang, S. Wang, J. Dong, CX3CL1/fractalkine enhances prostate cancer spinal metastasis by activating the Src/FAK pathway, Int. J. Oncol., 53 (2018), 1544–1556. https://doi.org/10.3892/ijo.2018.4487 doi: 10.3892/ijo.2018.4487
|
| [37] |
S. Chen, M. Huang, X. Hu, Interference with KCNJ2 inhibits proliferation, migration and EMT progression of apillary thyroid carcinoma cells by upregulating GNG2 expression, Mol. Med. Rep., 24 (2021), 1–10. https://doi.org/10.3892/mmr.2021.12261 doi: 10.3892/mmr.2021.12261
|
| [38] |
C. Ji, Y. Wang, D. Xiang, Q. Liu, Z. Zhou, F. Qian, et al., Kir2.1 interaction with Stk38 promotes invasion and metastasis of human gastric cancer by enhancing MEKK2-MEK1/2-ERK1/2 signaling, Cancer Res., 78 (2018), 3041–3053. https://doi.org/10.1158/0008-5472.CAN-17-3776 doi: 10.1158/0008-5472.CAN-17-3776
|
| [39] |
L. Liesenfeld, M. Kron, J. E. Gschwend, K. Herkommer, Prognostic factors for biochemical recurrence more than 10 years after radical prostatectomy, J. Urol., 197 (2017), 143–148. https://doi.org/10.1016/j.juro.2016.07.004 doi: 10.1016/j.juro.2016.07.004
|
| [40] |
C. R. Pound, A. W. Partin, M. A. Eisenberger, D. W. Chan, J. D. Pearson, P. C. Walsh, Natural history of progression after PSA elevation following radical prostatectomy, JAMA, 281 (1999), 1591–1597. https://doi.org/10.1001/jama.281.17.1591 doi: 10.1001/jama.281.17.1591
|
| [41] |
S. Shen, R. Zhang, Y. Jiang, Y. Li, L. Lin, Z. Liu, et al., Comprehensive analyses of m6A regulators and interactive coding and non-coding RNAs across 32 cancer types, Mol. Cancer, 20 (2021), 67. https://doi.org/10.1186/s12943-021-01362-2 doi: 10.1186/s12943-021-01362-2
|
| [42] |
T. Wu, Y. Dai, Tumor microenvironment and therapeutic response, Cancer Lett., 387 (2017), 61–68. https://doi.org/10.1016/j.canlet.2016.01.043 doi: 10.1016/j.canlet.2016.01.043
|
| [43] |
M. St Paul, P. S. Ohashi, The roles of CD8(+) T cell subsets in antitumor immunity, Trends Cell Biol., 30 (2020), 695–704. https://doi.org/10.1016/j.tcb.2020.06.003 doi: 10.1016/j.tcb.2020.06.003
|
| [44] |
M. Philip, A. Schietinger, CD8(+) T cell differentiation and dysfunction in cancer, Nat. Rev. Immunol., 22 (2022), 209–223. https://doi.org/10.1038/s41577-021-00574-3 doi: 10.1038/s41577-021-00574-3
|
| [45] |
J. Qiao, Z. Liu, C. Dong, Y. Luan, A. Zhang, C. Moore, et al., Targeting tumors with IL-10 prevents dendritic cell-mediated CD8(+) T cell apoptosis, Cancer Cell, 35 (2019), 901–915. https://doi.org/10.1016/j.ccell.2019.05.005 doi: 10.1016/j.ccell.2019.05.005
|
| [46] |
G. M. Lynn, C. Sedlik, F. Baharom, Y. Zhu, R. A. Ramirez-Valdez, V. L. Coble, et al., Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens, Nat. Biotechnol., 38 (2020), 320–332. https://doi.org/10.1038/s41587-019-0390-x doi: 10.1038/s41587-019-0390-x
|
| [47] |
S. Wang, W. Su, C. Zhong, T. Yang, W. Chen, G. Chen, et al., An eight-circRNA assessment model for predicting biochemical recurrence in prostate cancer, Front. Cell Dev. Biol., 8 (2020), 599494. https://doi.org/10.3389/fcell.2020.599494 doi: 10.3389/fcell.2020.599494
|
| [48] |
Y. Yanai, T. Kosaka, S. Mikami, H. Hongo, Y. Yasumizu, T. Takeda, et al., CD8-positive T cells and CD204-positive M2-like macrophages predict postoperative prognosis of very high-risk prostate cancer, Sci. Rep., 11 (2021), 22495. https://doi.org/10.1038/s41598-021-01900-4 doi: 10.1038/s41598-021-01900-4
|
| [49] |
J. R. Lees, B. Charbonneau, J. D. Hayball, K. Diener, M. Brown, R. Matusik, et al., T-cell recognition of a prostate specific antigen is not sufficient to induce prostate tissue destruction, Prostate, 66 (2006), 578–590. https://doi.org/10.1002/pros.20307 doi: 10.1002/pros.20307
|
| [50] |
V. R. de Porras, J. C. Pardo, L. Notario, O. Etxaniz, A. Font, Immune checkpoint inhibitors: A promising treatment option for metastatic castration-resistant prostate cancer, Int. J. Mol. Sci., 22 (2021), 4712. https://doi.org/10.3390/ijms22094712 doi: 10.3390/ijms22094712
|
| [51] |
M. Bilusic, R. A. Madan, J. L. Gulley, Immunotherapy of prostate cancer: Facts and hopes, Clin. Cancer Res., 23 (2017), 6764–6770. https://doi.org/10.1158/1078-0432.CCR-17-0019 doi: 10.1158/1078-0432.CCR-17-0019
|
| [52] |
E. S. Antonarakis, J. M. Piulats, M. Gross-Goupil, J. Goh, K. Ojamaa, C. J. Hoimes, et al., Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: Multicohort, open-label phase Ⅱ KEYNOTE-199 study, J. Clin. Oncol., 38 (2020), 395–405. https://doi.org/10.1200/JCO.19.01638 doi: 10.1200/JCO.19.01638
|
| [53] |
A. R. Hansen, C. Massard, P. A. Ott, N. B. Haas, J. S. Lopez, S. Ejadi, et al., Pembrolizumab for advanced prostate adenocarcinoma: Findings of the KEYNOTE-028 study, Ann. Oncol., 29 (2018), 1807–1813. https://doi.org/10.1093/annonc/mdy232 doi: 10.1093/annonc/mdy232
|
| [54] |
R. Mo, Z. Han, Y. Liang, J. Ye, S. Wu, S. X. Lin, et al., Expression of PD-L1 in tumor-associated nerves correlates with reduced CD8(+) tumor-associated lymphocytes and poor prognosis in prostate cancer, Int. J. Cancer, 144 (2019), 3099–3110. https://doi.org/10.1002/ijc.32061 doi: 10.1002/ijc.32061
|
| [55] |
F. Petitprez, N. Fossati, Y. Vano, M. Freschi, E. Becht, R. Lucianò, et al., PD-L1 expression and CD8(+) T-cell infiltrate are associated with clinical progression in patients with node-positive prostate cancer, Eur. Urol. Focus, 5 (2019), 192–196. https://doi.org/10.1016/j.euf.2017.05.013 doi: 10.1016/j.euf.2017.05.013
|
| [56] |
C. Vicier, P. Ravi, L. Kwak, L. Werner, Y. Huang, C. Evan, et al., Association between CD8 and PD-L1 expression and outcomes after radical prostatectomy for localized prostate cancer, Prostate, 81 (2021), 50–57. https://doi.org/10.1002/pros.24079 doi: 10.1002/pros.24079
|
| [57] |
H. Gevensleben, D. Dietrich, C. Golletz, S. Steiner, M. Jung, T. Thiesler, et al., The immune checkpoint regulator PD-L1 is highly expressed in aggressive primary prostate cancer, Clin. Cancer Res., 22 (2016), 1969–1977. https://doi.org/10.1158/1078-0432.CCR-15-2042 doi: 10.1158/1078-0432.CCR-15-2042
|
| [58] |
B. T. Rekoske, B. M. Olson, D. G. McNeel, Antitumor vaccination of prostate cancer patients elicits PD-1/PD-L1 regulated antigen-specific immune responses, Oncoimmunology, 5 (2016), e1165377. https://doi.org/10.1080/2162402X.2016.1165377 doi: 10.1080/2162402X.2016.1165377
|
 mbe-19-11-528-Supplementary tables.zip mbe-19-11-528-Supplementary tables.zip |
 |
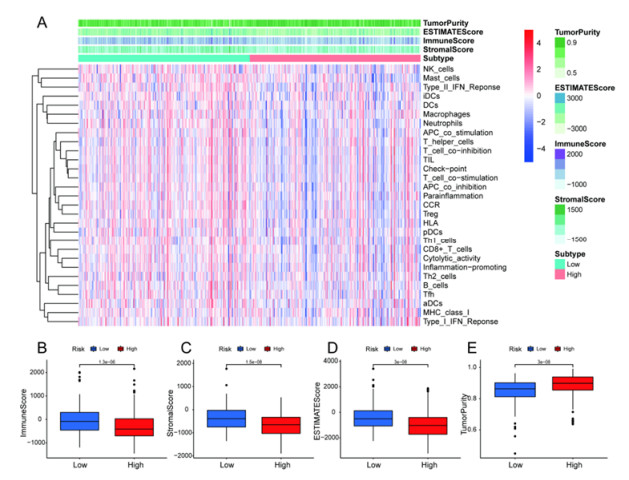
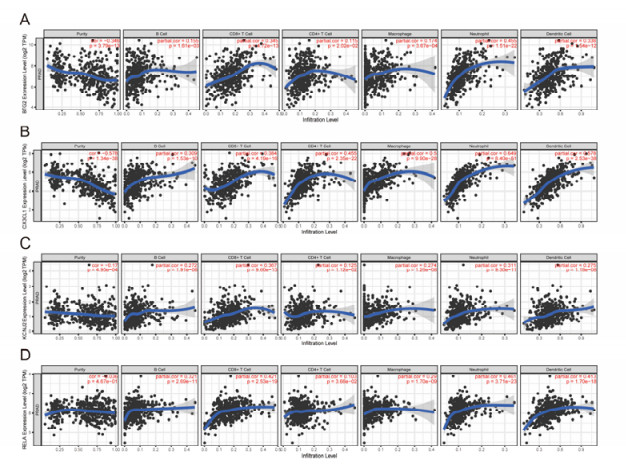
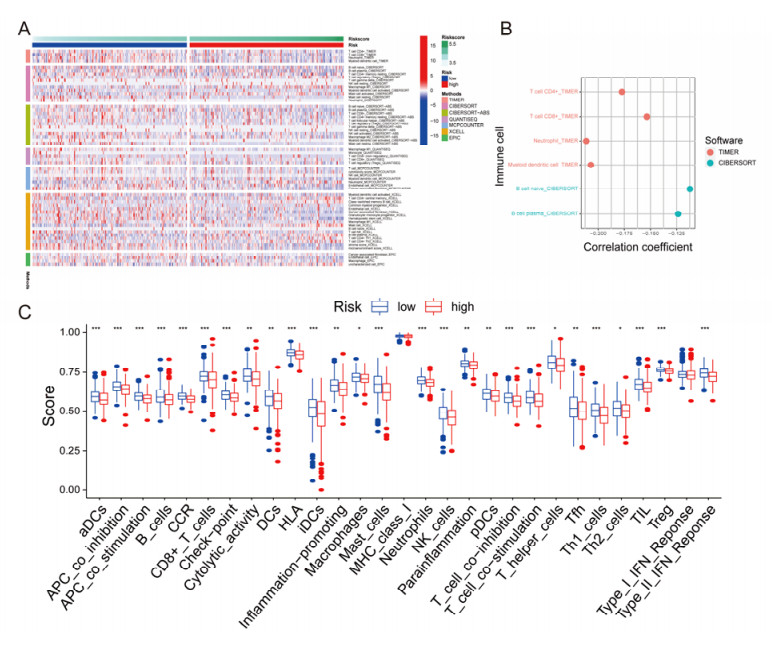
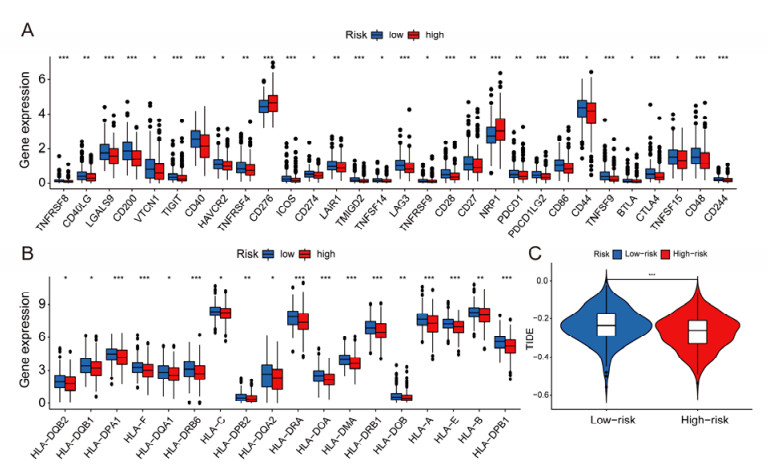
Figures(13)

Yong Luo, Xiaopeng Liu, Jingbo Lin, Weide Zhong, Qingbiao Chen. Development and validation of novel inflammatory response-related gene signature to predict prostate cancer recurrence and response to immune checkpoint therapy[J]. Mathematical Biosciences and Engineering, 2022, 19(11): 11345-11366. doi: 10.3934/mbe.2022528







 DownLoad:
DownLoad: