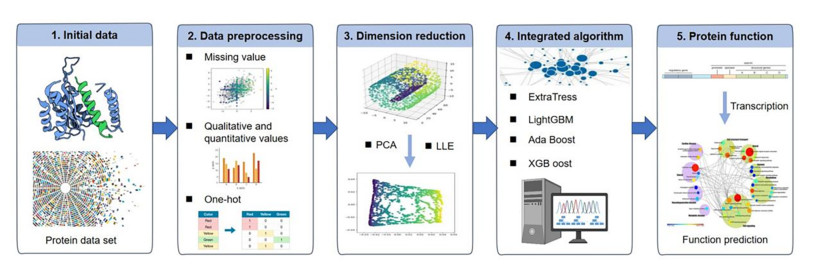
Protein interactions are the foundation of all metabolic activities of cells, such as apoptosis, the immune response, and metabolic pathways. In order to optimize the performance of protein interaction prediction, a coding method based on normalized difference sequence characteristics (NDSF) of amino acid sequences is proposed. By using the positional relationships between amino acids in the sequences and the correlation characteristics between sequence pairs, NDSF is jointly encoded. Using principal component analysis (PCA) and local linear embedding (LLE) dimensionality reduction methods, the coded 174-dimensional human protein sequence vector is extracted using sequence features. This study compares the classification performance of four ensemble learning methods (AdaBoost, Extra trees, LightGBM, XGBoost) applied to PCA and LLE features. Cross-validation and grid search methods are used to find the best combination of parameters. The results show that the accuracy of NDSF is generally higher than that of the sequence matrix-based coding method (MOS) coding method, and the loss and coding time can be greatly reduced. The bar chart of feature extraction shows that the classification accuracy is significantly higher when using the linear dimensionality reduction method, PCA, compared to the nonlinear dimensionality reduction method, LLE. After classification with XGBoost, the model accuracy reaches 99.2%, which provides the best performance among all models. This study suggests that NDSF combined with PCA and XGBoost may be an effective strategy for classifying different human protein interactions.
Citation: Xiaoman Zhao, Xue Wang, Zhou Jin, Rujing Wang. A normalized differential sequence feature encoding method based on amino acid sequences[J]. Mathematical Biosciences and Engineering, 2023, 20(8): 14734-14755. doi: 10.3934/mbe.2023659
Protein interactions are the foundation of all metabolic activities of cells, such as apoptosis, the immune response, and metabolic pathways. In order to optimize the performance of protein interaction prediction, a coding method based on normalized difference sequence characteristics (NDSF) of amino acid sequences is proposed. By using the positional relationships between amino acids in the sequences and the correlation characteristics between sequence pairs, NDSF is jointly encoded. Using principal component analysis (PCA) and local linear embedding (LLE) dimensionality reduction methods, the coded 174-dimensional human protein sequence vector is extracted using sequence features. This study compares the classification performance of four ensemble learning methods (AdaBoost, Extra trees, LightGBM, XGBoost) applied to PCA and LLE features. Cross-validation and grid search methods are used to find the best combination of parameters. The results show that the accuracy of NDSF is generally higher than that of the sequence matrix-based coding method (MOS) coding method, and the loss and coding time can be greatly reduced. The bar chart of feature extraction shows that the classification accuracy is significantly higher when using the linear dimensionality reduction method, PCA, compared to the nonlinear dimensionality reduction method, LLE. After classification with XGBoost, the model accuracy reaches 99.2%, which provides the best performance among all models. This study suggests that NDSF combined with PCA and XGBoost may be an effective strategy for classifying different human protein interactions.

| [1] |
C. Gustafsson, J. Minshull, S. Govindarajan, J. Ness, A. Villalobos, Engineering genes for predictable protein expression, Protein Expression Purif., 83 (2012), 37–46. https://dx.doi.org/10.1016/j.pep.2012.02.013 doi: 10.1016/j.pep.2012.02.013

|
| [2] |
L. Y. Mei, M. R. Montoya, G. M. Quanrud, M. Tran, A. Villa-Sharma, M. Huang, et al., Bait correlation improves interactor identification by tandem mass tag-affinity Purification-Mass spectrometry, J. Proteome Res., 19 (2020), 1565–1573. https://dx.doi.org/10.1021/acs.jproteome.9b00825 doi: 10.1021/acs.jproteome.9b00825
|
| [3] |
I. Paspaltsis, E. Kesidou, O. Touloumi, R. Lagoudaki, M. Boziki, M. Samiotaki, et al., Application of antibody phage display to identify potential antigenic neural precursor cell proteins, J. Biol. Res. Thessaloniki, 27 (2020). https://dx.doi.org/10.1186/s40709-020-00123-4 doi: 10.1186/s40709-020-00123-4
|
| [4] |
A. Rami, M. Behdani, N. Yardehnavi, M. Habibi-Anbouhi, F. Kazemi-Lomedasht, An overview on application of phage display technique in immunological studies, Asian Pac. J. Trop. Biomed., 7 (2017), 599–602. https://dx.doi.org/10.1016/j.apjtb.2017.06.001 doi: 10.1016/j.apjtb.2017.06.001
|
| [5] |
S. Schuette, B. Piatkowski, A. Corley, D. Lang, M. Geisler, Predicted protein-protein interactions in the moss Physcomitrella patens: a new bioinformatic resource, BMC Bioinf., 16 (2015). https://dx.doi.org/10.1186/s12859-015-0524-1 doi: 10.1186/s12859-015-0524-1
|
| [6] |
L. L. Song, S. B. Ning, J. X. Hou, Y. Zhao, Performance of protein-ligand docking with CDK4/6 inhibitors: a case study, Math. Biosci. Eng., 18 (2020), 456–470. https://dx.doi.org/10.3934/mbe.2021025 doi: 10.3934/mbe.2021025
|
| [7] |
Y. C. Wang, J. G. Wang, Z. X. Yang, N. Deng, Sequence-based protein-protein interaction prediction via support vector machine, J. Syst. Sci. Complexity, 23 (2010), 1012–1023. https://dx.doi.org/10.1007/s11424-010-0214-z doi: 10.1007/s11424-010-0214-z
|
| [8] |
L. Yang, X. D. Zhao, X. L. Tang, Predicting disease-related proteins based on clique backbone in protein-protein interaction network, Int. J. Biol. Sci., 10 (2014), 677–688. https://dx.doi.org/10.7150/ijbs.8430 doi: 10.7150/ijbs.8430
|
| [9] |
H. P. Zhang, L. B. Liao, K. M. Saravanan, P. Yin, Y. Wei, DeepBindRG: a deep learning based method for estimating effective protein-ligand affinity, PeerJ, 7 (2019). https://dx.doi.org/10.7717/peerj.7362 doi: 10.7717/peerj.7362
|
| [10] |
X. Y. Zhou, I. Naguro, H. Ichijo, K. Watanabe, Mitogen-activated protein kinases as key players in osmotic stress signaling, Biochim. Biophys. Acta Gen. Subj., 1860 (2016), 2037–2052. https://dx.doi.org/10.1016/j.bbagen.2016.05.032 doi: 10.1016/j.bbagen.2016.05.032
|
| [11] | Y. Z. Zhou, Y. Gao, Y. Y. Zheng, Prediction of protein-protein interactions using local description of amino acid sequence, in Advances in Computer Science and Education Applications, Springer, 2011. https://doi.org/10.1007/978-3-642-22456-0_37 |
| [12] |
Y. H. Zhu, X. R. Zhang, S. J. Xie, W. Bao, J. Chen, Q. Wu, et al., Oxidative phosphorylation regulates interleukin-10 production in regulatory B cells via the extracellular signal-related kinase pathway, Immunology, 167 (2022), 576–589. https://dx.doi.org/10.1111/imm.13554 doi: 10.1111/imm.13554
|
| [13] |
X. Cao, G. X. Yu, W. Ren, M. Guo, J. Wang, DualWMDR: Detecting epistatic interaction with dual screening and multifactor dimensionality reduction, Hum. Mutat., 41 (2020), 719–734. https://dx.doi.org/10.1002/humu.23951 doi: 10.1002/humu.23951
|
| [14] |
P. Malvi, R. Janostiak, S. Chava, P. Manrai, E. Yoon, K. Singh, et al., LIMK2 promotes the metastatic progression of triple-negative breast cancer by activating SRPK1, Oncogenesis, 9 (2020). https://dx.doi.org/10.1038/s41389-020-00263-1 doi: 10.1038/s41389-020-00263-1
|
| [15] |
Y. M. Wu, M. Zhou, K. Chen, S. Chen, X. Xiao, Z. Ji, et al., Alkali-metal hexamethyldisilazide initiated polymerization on alpha-amino acid N-substituted N-carboxyanhydrides for facile polypeptoid synthesis, Chin. Chem. Lett., 32 (2021), 1675–1678. https://dx.doi.org/10.1016/j.cclet.2021.02.039 doi: 10.1016/j.cclet.2021.02.039
|
| [16] |
W. Zhang, X. L. Xue, C. W. Xie, Y. Li, J. Liu, H. Chen, et al., CEGSO: Boosting essential proteins prediction by integrating protein complex, gene expression, gene ontology, subcellular localization and Orthology information, Interdiscip. Sci.-Comput. Life Sci., 13 (2021), 349–361. https://dx.doi.org/10.1007/s12539-021-00426-7 doi: 10.1007/s12539-021-00426-7
|
| [17] |
Y. N. Shen, Y. J. Ding, J. J. Tang, Q. Zou, F. Guo, Critical evaluation of web-based prediction tools for human protein subcellular localization, Briefings Bioinf. 21 (2020), 1628–1640. https://dx.doi.org/10.1093/bib/bbz106 doi: 10.1093/bib/bbz106
|
| [18] |
T. Z. Yu, W. S. Zhang, Semisupervised multilabel learning with joint dimensionality reduction, IEEE Signal Process Lett., 23 (2016), 795–799. https://dx.doi.org/10.1109/lsp.2016.2554361 doi: 10.1109/lsp.2016.2554361
|
| [19] |
C. Chen, Q. M. Zhang, Q. Ma, B. Yu, LightGBM-PPI: Predicting protein-protein interactions through LightGBM with multi-information fusion, Chemom. Intell. Lab. Syst., 191 (2019), 54–64. https://dx.doi.org/10.1016/j.chemolab.2019.06.003 doi: 10.1016/j.chemolab.2019.06.003
|
| [20] |
P. P. Hao, H. Li, L. Zhou, H. Sun, J. Han, Z. Zhang, Serum metal ion-induced cross-linking of photoelectrochemical peptides and circulating proteins for evaluating cardiac ischemia/reperfusion, ACS Sens., 7 (2022), 775–783. https://dx.doi.org/10.1021/acssensors.1c02305 doi: 10.1021/acssensors.1c02305
|
| [21] |
D. J. W. Tay, Z. Z. R. Lew, J. J. H. Chu, K. S. Tan, Uncovering novel viral innate immune evasion strategies: What has SARS-CoV-2 taught us, Front. Microbiol., 13 (2022). https://dx.doi.org/10.3389/fmicb.2022.844447 doi: 10.3389/fmicb.2022.844447
|
| [22] |
K. Y. Huang, Q. H. Fang, W. M. Sun, S. He, Q. Yao, J. Xie, et al., Cucurbit[n]uril supramolecular assemblies-regulated charge transfer for luminescence switching of gold nanoclusters, J. Phys. Chem. Lett., 13 (2022), 419–426. https://dx.doi.org/10.1021/acs.jpclett.1c03917 doi: 10.1021/acs.jpclett.1c03917
|
| [23] |
Z. Y. Wu, H. Yin, H. He, Y. Li, Dynamic-LSTM hybrid models to improve seasonal drought predictions over China, J. Hydrol., 615 (2022). https://dx.doi.org/10.1016/j.jhydrol.2022.128706 doi: 10.1016/j.jhydrol.2022.128706
|
| [24] |
C. G. Yan, L. X. Meng, L. Li, J. Zhang, Z. Wang, J. Yin, et al., Age-invariant face recognition by multi-feature fusion and decomposition with self-attention, ACM Trans. Multimedia Comput. Commun. Appl., 18 (2022). https://dx.doi.org/10.1145/3472810 doi: 10.1145/3472810
|
| [25] |
W. Wang, D. S. Tekcham, M. Yan, Z. Wang, H. Qi, X. Liu, et al., Biochemical reactions in metabolite-protein interaction, Chin. Chem. Lett., 29 (2018), 645–647. https://dx.doi.org/10.1016/j.cclet.2017.10.002 doi: 10.1016/j.cclet.2017.10.002
|
| [26] |
Y. D. Liang, R. F. Sun, L. J. Li, F. Yuan, W. Liang, L. Wang, et al., A functional polymorphism in the promoter of MiR-143/145 is associated with the risk of cervical squamous cell carcinoma in Chinese women a case-control study, Medicine, 94 (2015). https://dx.doi.org/10.1097/MD.0000000000001289 doi: 10.1097/MD.0000000000001289
|
| [27] |
M. Braaksma, E. S. Martens-Uzunova, P. J. Punt, P. J. Schaap, An inventory of the Aspergillus niger secretome by combining in silico predictions with shotgun proteomics data, BMC Genomics, 11 (2010). https://dx.doi.org/10.1186/1471-2164-11-584 doi: 10.1186/1471-2164-11-584
|
| [28] |
P. Walther, A. Krauss, S. Naumann, Lewis pair polymerization of epoxides via zwitterionic species as a route to High-Molar-Mass polyethers, Angew. Chem. Int. Ed., 58 (2019), 10737–10741. https://dx.doi.org/10.1002/anie.201904806 doi: 10.1002/anie.201904806
|
| [29] |
Y. M. Wu, D. F. Zhang, P. C. Ma, R. Zhou, L. Hua, R. Liu, Lithium hexamethyldisilazide initiated superfast ring opening polymerization of alpha-amino acid N-carboxyanhydrides, Nat. Commun., 9 (2018). https://dx.doi.org/10.1038/s41467-018-07711-y doi: 10.1038/s41467-018-07711-y
|
| [30] |
C. H. Xin, X. F. Ban, Z. B. Gu, C. Li, L. Cheng, Y. Hong, et al., Non-classical secretion of 1, 4-alpha-glucan branching enzymes without signal peptides in Escherichia coli, Int. J. Biol. Macromol., 132 (2019), 759–765. https://dx.doi.org/10.1016/j.ijbiomac.2019.04.002 doi: 10.1016/j.ijbiomac.2019.04.002
|
| [31] |
Y. J. Zhang, S. Yu, R. P. Xie, J. Li, A. Leier, T. Marquez-Lago, et al., PeNGaRoo, a combined gradient boosting and ensemble learning framework for predicting non-classical secreted proteins, Bioinformatics, 36 (2020), 704–712. https://dx.doi.org/10.1093/bioinformatics/btz629 doi: 10.1093/bioinformatics/btz629
|
| [32] |
C. J. Fee, J. A. Van, Alstine PEG-proteins: Reaction engineering and separation issues, Chem. Eng. Sci., 61 (2006), 924–939. https://dx.doi.org/10.1016/j.ces.2005.04.040 doi: 10.1016/j.ces.2005.04.040
|
| [33] |
C. H. Hung, H. L. Huang, K. T. Hsu, S. J. Ho, S. Y. Ho, Prediction of non-classical secreted proteins using informative physicochemical properties, Interdiscip. Sci.: Comput. Life Sci., 2 (2010), 263–270. https://dx.doi.org/10.1007/s12539-010-0023-z doi: 10.1007/s12539-010-0023-z
|
| [34] |
A. X. Wang, S. S. Chukova, B. P. Nguyen, Ensemble k-nearest neighbors based on centroid displacement, Inf. Sci., 629 (2023), 313–323. https://dx.doi.org/10.1016/j.ins.2023.02.004 doi: 10.1016/j.ins.2023.02.004
|
| [35] |
B. P. Nguyen, W. L. Tay, C. K. Chui, Robust biometric recognition from palm depth images for gloved hands, IEEE Trans. Hum.-Mach. Syst., 45 (2015), 799–804. https://dx.doi.org/10.1109/THMS.2015.2453203 doi: 10.1109/THMS.2015.2453203
|
| [36] |
T. Wang, W. Wang, H. Liu, T. Li, Research on a face real-time tracking algorithm based on particle filter multi-feature fusion, Sensors, 19 (2019). https://dx.doi.org/10.3390/s19051245 doi: 10.3390/s19051245
|
| [37] |
H. J. Tao, X. B. Lu, Smoke vehicle detection based on multi-feature fusion and hidden Markov model, J. Real-Time Image Process., 17 (2020), 745–758. https://dx.doi.org/10.1007/s11554-019-00856-z doi: 10.1007/s11554-019-00856-z
|
| [38] |
A. Berg, O. Kukharenko, M. Scheffner, C. Peter, Towards a molecular basis of ubiquitin signaling: A dual-scale simulation study of ubiquitin dimers, PLOS Comput. Biol., 14 (2018). https://dx.doi.org/10.1371/journal.pcbi.1006589 doi: 10.1371/journal.pcbi.1006589
|
| [39] |
V. J. Jameson, T. Luke, Y. T. Yan, A. Hind, M. Evrard, K. Man, et al., Unlocking autofluorescence in the era of full spectrum analysis: Implications for immunophenotype discovery projects, Cytometry Part A, 101 (2022), 922–941. https://dx.doi.org/10.1002/cyto.a.24555 doi: 10.1002/cyto.a.24555
|
| [40] |
J. J. Zhang, S. Y. Wang, P. Zhang, S. Fan, H. Dai, Y. Xiao, et al., Engineering a cationic supramolecular charge switch for facile amino acids enantiodiscrimination based on extended-gate field effect transistors, Chin. Chem. Lett., 33 (2022), 3873–3878. https://dx.doi.org/10.1016/j.cclet.2021.11.081 doi: 10.1016/j.cclet.2021.11.081
|





Figures(6) / Tables(6)

Xiaoman Zhao, Xue Wang, Zhou Jin, Rujing Wang. A normalized differential sequence feature encoding method based on amino acid sequences[J]. Mathematical Biosciences and Engineering, 2023, 20(8): 14734-14755. doi: 10.3934/mbe.2023659







 DownLoad:
DownLoad: