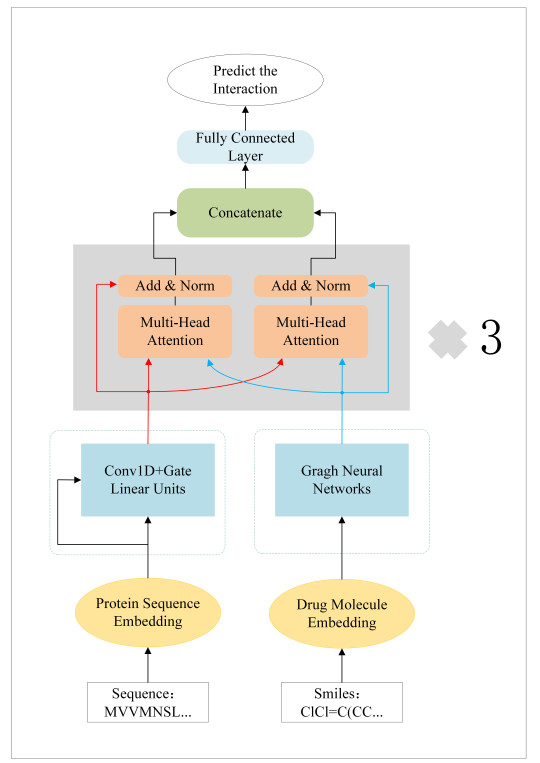
The prediction of drug-target protein interaction (DTI) is a crucial task in the development of new drugs in modern medicine. Accurately identifying DTI through computer simulations can significantly reduce development time and costs. In recent years, many sequence-based DTI prediction methods have been proposed, and introducing attention mechanisms has improved their forecasting performance. However, these methods have some shortcomings. For example, inappropriate dataset partitioning during data preprocessing can lead to overly optimistic prediction results. Additionally, only single non-covalent intermolecular interactions are considered in the DTI simulation, ignoring the complex interactions between their internal atoms and amino acids. In this paper, we propose a network model called Mutual-DTI that predicts DTI based on the interaction properties of sequences and a Transformer model. We use multi-head attention to extract the long-distance interdependent features of the sequence and introduce a module to extract the sequence's mutual interaction features in mining complex reaction processes of atoms and amino acids. We evaluate the experiments on two benchmark datasets, and the results show that Mutual-DTI outperforms the latest baseline significantly. In addition, we conduct ablation experiments on a label-inversion dataset that is split more rigorously. The results show that there is a significant improvement in the evaluation metrics after introducing the extracted sequence interaction feature module. This suggests that Mutual-DTI may contribute to modern medical drug development research. The experimental results show the effectiveness of our approach. The code for Mutual-DTI can be downloaded from https://github.com/a610lab/Mutual-DTI.
Citation: Jiahui Wen, Haitao Gan, Zhi Yang, Ran Zhou, Jing Zhao, Zhiwei Ye. Mutual-DTI: A mutual interaction feature-based neural network for drug-target protein interaction prediction[J]. Mathematical Biosciences and Engineering, 2023, 20(6): 10610-10625. doi: 10.3934/mbe.2023469
The prediction of drug-target protein interaction (DTI) is a crucial task in the development of new drugs in modern medicine. Accurately identifying DTI through computer simulations can significantly reduce development time and costs. In recent years, many sequence-based DTI prediction methods have been proposed, and introducing attention mechanisms has improved their forecasting performance. However, these methods have some shortcomings. For example, inappropriate dataset partitioning during data preprocessing can lead to overly optimistic prediction results. Additionally, only single non-covalent intermolecular interactions are considered in the DTI simulation, ignoring the complex interactions between their internal atoms and amino acids. In this paper, we propose a network model called Mutual-DTI that predicts DTI based on the interaction properties of sequences and a Transformer model. We use multi-head attention to extract the long-distance interdependent features of the sequence and introduce a module to extract the sequence's mutual interaction features in mining complex reaction processes of atoms and amino acids. We evaluate the experiments on two benchmark datasets, and the results show that Mutual-DTI outperforms the latest baseline significantly. In addition, we conduct ablation experiments on a label-inversion dataset that is split more rigorously. The results show that there is a significant improvement in the evaluation metrics after introducing the extracted sequence interaction feature module. This suggests that Mutual-DTI may contribute to modern medical drug development research. The experimental results show the effectiveness of our approach. The code for Mutual-DTI can be downloaded from https://github.com/a610lab/Mutual-DTI.

| [1] |
C. Chen, H. Shi, Y. Han, Z. Jiang, X. Cui, B. Yu, DNN-DTIs: Improved drug-target interactions prediction using XGBoost feature selection and deep neural network, Comput. Biol. Med., 136 (2021), 104676. https://doi.org/10.1101/2020.08.11.247437 doi: 10.1101/2020.08.11.247437

|
| [2] |
X. Ru, X. Ye, T. Sakurai, Q. Zou, C. Lin, Current status and future prospects of drug–target interaction prediction, Briefings Funct. Genomics, 20 (2021), 312–322. https://doi.org/10.1093/bfgp/elab031 doi: 10.1093/bfgp/elab031
|
| [3] |
G. Huang, F. Yan, D. Tan, A review of computational methods for predicting drug targets, Curr. Protein Pept. Sci., 19 (2018), 562–572. https://doi.org/10.1016/j.jad.2018.12.111 doi: 10.1016/j.jad.2018.12.111
|
| [4] |
J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, et al., Applications of machine learning in drug discovery and development, Nat. Rev. Drug Discovery, 18 (2019), 463–477. https://doi.org/10.1038/s41573-019-0024-5 doi: 10.1038/s41573-019-0024-5
|
| [5] |
E. Maia, L. C. Assis, T. Oliveira, A.M. de Silva, A. G. Taranto, Structure-based virtual screening: from classical to artificial intelligence, Front. Chem., 8 (2020), 343. https://doi.org/10.3389/fchem.2020.00343 doi: 10.3389/fchem.2020.00343
|
| [6] |
H. Mubarak, S. Naomie, A. D. Mohammed, S. Faisal, A. Ali, Adapting document similarity measures for ligand-based virtual screening, Molecules, 21 (2016), 476. https://doi.org/10.3390/molecules21040476 doi: 10.3390/molecules21040476
|
| [7] |
R. Ferdousi, R. Safdari, Y. Omidi, Computational prediction of drug-drug interactions based on drugs functional similarities, J. Biomed. Inf., 70 (2017), 54. https://doi.org/10.1016/j.jbi.2017.04.021 doi: 10.1016/j.jbi.2017.04.021
|
| [8] |
M. Bredel, E. Jacoby, Chemogenomics: An emerging strategy for rapid target and drug discovery, Nat. Rev. Genet., 5 (2004), 262–275. https://doi.org/10.1038/nrg1317 doi: 10.1038/nrg1317
|
| [9] |
K. Bleakley, Y. Yamanishi, Supervised prediction of drug–target interactions using bipartite local models, Bioinformatics, 25 (2009), 2397–2403. https://doi.org/10.1093/bioinformatics/btp433 doi: 10.1093/bioinformatics/btp433
|
| [10] |
F. Cheng, Y. Zhou, J. Li, W. Li, G. Liu, Y. Tang, Prediction of chemical–protein interactions: multitarget-QSAR versus computational chemogenomic methods, Mol. BioSyst., 8 (2012), 2373–2384. https://doi.org/10.1039/C2MB25110H doi: 10.1039/C2MB25110H
|
| [11] |
M. Gönen, Predicting drug–target interactions from chemical and genomic kernels using Bayesian matrix factorization, Bioinformatics, 28 (2012), 2304–2310. https://doi.org/10.1093/bioinformatics/bts360 doi: 10.1093/bioinformatics/bts360
|
| [12] |
S. Liu, J. An, J. Zhao, S. Zhao, H. Lv, S. Wang, et al., Drug-Target interaction prediction based on multisource information weighted fusion, Contrast Media Mol. Imaging, 2021 (2021). https://doi.org/10.1155/2021/6044256 doi: 10.1155/2021/6044256
|
| [13] |
J. Li, X. Yang, Y. Guan, Z. Pan, Prediction of drug–target interaction using dual-network integrated logistic matrix factorization and knowledge graph embedding, Molecules, 27 (2022), 5131. https://doi.org/10.3390/molecules27165131 doi: 10.3390/molecules27165131
|
| [14] |
Y. Ding, J. Tang, F. Guo, Q. Zou, Identification of drug–target interactions via multiple kernel-based triple collaborative matrix factorization, Brief. Bioinf., 23 (2022). https://doi.org/10.1093/bib/bbab582 doi: 10.1093/bib/bbab582
|
| [15] |
L. Jacob, J. P. Vert, Protein-ligand interaction prediction: An improved chemogenomics approach, Bioinformatics, 24 (2008), 2149–2156. https://doi.org/10.1093/bioinformatics/btn409 doi: 10.1093/bioinformatics/btn409
|
| [16] |
T. Van Laarhoven, S. B Nabuurs, E. Marchiori, Gaussian interaction profile kernels for predicting drug–target interaction, Bioinformatics, 27 (2011), 3036–3043. https://doi.org/10.1093/bioinformatics/btr500 doi: 10.1093/bioinformatics/btr500
|
| [17] |
F. Wang, D. Liu, H. Wang, C. Luo, M. Zheng, H. Liu, et al., Computational screening for active compounds targeting protein sequences: Methodology and experimental validation, J. Chem. Inf. Model., 51 (2011), 2821–2828. https://doi.org/10.1021/ci200264h doi: 10.1021/ci200264h
|
| [18] |
Y. Wang, J. Zeng, Predicting drug-target interactions using restricted Boltzmann machines, Bioinformatics, 29 (2013), i126–i134. https://doi.org/10.1093/bioinformatics/btt234 doi: 10.1093/bioinformatics/btt234
|
| [19] |
Y. Yamanishi, M. Araki, A. Gutteridge, W. Honda, M. Kanehisa, Prediction of drug–target interaction networks from the integration of chemical and genomic spaces, Bioinformatics, 24 (2008), i232–i240. https://doi.org/10.1093/bioinformatics/btn162 doi: 10.1093/bioinformatics/btn162
|
| [20] |
K. Tian, M. Shao, Y. Wang, J. Guan, S. Zhou, Boosting compound-protein interaction prediction by deep learning, Methods, 110 (2016), 64–72. https://doi.org/10.1016/j.ymeth.2016.06.024 doi: 10.1016/j.ymeth.2016.06.024
|
| [21] |
I. Lee, J. Keum, H. Nam, DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences, PLoS Comput. Biol., 15 (2019), e1007129. https://doi.org/10.1371/journal.pcbi.1007129 doi: 10.1371/journal.pcbi.1007129
|
| [22] |
M. Tsubaki, K. Tomii, J. Sese, Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences, Bioinformatics, 35 (2019), 309–318. https://doi.org/10.1093/bioinformatics/bty535 doi: 10.1093/bioinformatics/bty535
|
| [23] |
S. Li, F. Wan, H. Shu, T. Jiang, D. Zhao, J. Zeng, MONN: A multi-objective neural network for predicting compound-protein interactions and affinities, Cell Syst., 10 (2020), 308–322. https://doi.org/10.1016/j.cels.2020.03.002 doi: 10.1016/j.cels.2020.03.002
|
| [24] |
R. Zamora-Resendiz, S. Crivelli, Structural learning of proteins using graph convolutional neural networks, BioRxiv, (2019), 610444. https://doi.org/10.1101/610444 doi: 10.1101/610444
|
| [25] | S. Ryu, J. Lim, S. H. Hong, W. Y. Kim, Deeply learning molecular structure-property relationships using attention-and gate-augmented graph convolutional network, preprint, arXiv: 1805.10988. https://doi.org/10.48550/arXiv.1805.10988 |
| [26] |
X. Ru, X. Ye, T. Sakurai, Q. Zou, NerLTR-DTA: Drug–target binding affinity prediction based on neighbor relationship and learning to rank, Bioinformatics, 38 (2022), 1964–1971. https://doi.org/10.1093/bioinformatics/btac048 doi: 10.1093/bioinformatics/btac048
|
| [27] | J. Wang, X. Li, H. Zhang, GNN-PT: Enhanced prediction of compound-protein interactions by integrating protein transformer, preprint, arXiv: 2009.00805. https://doi.org/10.48550/arXiv.2009.00805 |
| [28] |
L. Chen, X. Tan, D. Wang, F. Zhong, X. Liu, T. Yang, et al., TransformerCPI: Improving compound–protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments, Bioinformatics, 36 (2020), 4406–4414. https://doi.org/10.1093/bioinformatics/btaa524 doi: 10.1093/bioinformatics/btaa524
|
| [29] |
Z. H. Ren, Z. H. You, Q. Zou, C. Q. Yu, Y. F. Ma, Y. J. Guan, et al., DeepMPF: Deep learning framework for predicting drug–target interactions based on multi-modal representation with meta-path semantic analysis, J. Transl. Med., 21 (2023), 1–18. https://doi.org/10.1186/s12967-023-03876-3 doi: 10.1186/s12967-023-03876-3
|
| [30] | Y. Wu, L. Zhu, Y. Yan, Y. Yang, Dual attention matching for audio-visual event localization, in Proceedings of the IEEE/CVF International Conference on Computer Vision, (2019), 6292–6300. |
| [31] | F. Costa, K. De Grave, Fast neighborhood subgraph pairwise distance kernel, in Proceedings of the 26th International Conference on Machine Learning, (2010), 255–262. |
| [32] | Y. N. Dauphin, A. Fan, M. Auli, D. Grangier, Language modeling with gated convolutional networks, in International Conference on Machine Learning, (2017), 933–941. |
| [33] | D. P. Kingma, J. Ba, Adam: A method for stochastic optimization, preprint, arXiv: 1412.6980. https://doi.org/10.48550/arXiv.1412.6980 |
| [34] |
H. Liu, J. Sun, J. Guan, J. Zheng, S. Zhou, Improving compound–protein interaction prediction by building up highly credible negative samples, Bioinformatics, 31 (2015), i221–i229. https://doi.org/10.1093/bioinformatics/btv256 doi: 10.1093/bioinformatics/btv256
|
| [35] |
Q. Zhao, H. Zhao, K. Zheng, J. Wang, HyperAttentionDTI: Improving drug–protein interaction prediction by sequence-based deep learning with attention mechanism, Bioinformatics, 38 (2022), 655–662. https://doi.org/10.1093/bioinformatics/btab715 doi: 10.1093/bioinformatics/btab715
|



Figures(4) / Tables(7)

Jiahui Wen, Haitao Gan, Zhi Yang, Ran Zhou, Jing Zhao, Zhiwei Ye. Mutual-DTI: A mutual interaction feature-based neural network for drug-target protein interaction prediction[J]. Mathematical Biosciences and Engineering, 2023, 20(6): 10610-10625. doi: 10.3934/mbe.2023469







 DownLoad:
DownLoad: