Citation: Hanyu Zhao, Chao Che, Bo Jin, Xiaopeng Wei. A viral protein identifying framework based on temporal convolutional network[J]. Mathematical Biosciences and Engineering, 2019, 16(3): 1709-1717. doi: 10.3934/mbe.2019081

| [1] | O. P. Zhirnov, A. L. Ksenofontov and N. D. Klenk, Influenza A virus M1 matrix protein is similar to protease inhibitors, Dokl. Akad. Nauk, 367(1999), 690–693. |
| [2] | S. Niu, T. Huang and K. Feng, et al., Prediction of tyrosine sulfation with mRMR feature selection and analysis, J. Proteome Res., 9(2010), 6490–6497. |
| [3] | S. Bai, J. Z. Kolter, and V. Koltun, An Empirical Evaluation of Generic Convolutional and Recurrent Networks for Sequence Modeling, preprint, arXiv:0707.0078. |
| [4] | Z. C. Lipton, J. Berkowitz, and C. Elkan, A critical review of recurrent neural networks for sequence learning, preprint, arXiv:1506.00019. |
| [5] | J. H. Friedman, Greedy function approximation: A gradient boosting machine, Ann. Stat., 29(2001), 1189–1232. |
| [6] | D. Wang, N. K. Lee and T. S. Dillon, et al., Protein sequences classification using radial basis function (RBF) neural networks, In: International Conference on Neural Information Processing, 2(2002), 764–768. |
| [7] | C. Lin, Y. Zou and J. Qin, et al., Hierarchical Classification of Protein Folds Using a Novel Ensemble Classifier, PLoS One, 8(2013), e56499. |
| [8] | T. K. Lee and T. Nguyen, Protein Family Classication with Neural Networks, Stanford University, 2016, Available online: https://cs224d.stanford.edu/reports/LeeNguyen.pdf. |
| [9] | H. Li, H. Yu and X. Gong, A Deep Learning Model for Predicting RNA-Binding Proteins Only from Primary Sequences, J. Comput. Res. Dev., 55(2018), 93–101. |
| [10] | N. V. Chawla, K. W. Bowyer and L. O. Hall, et al., SMOTE: synthetic minority over-sampling technique, J. Artif. Intell. Res., 16(2002), 321-357. |
| [11] | W. Fan, S. J. Stolfo and J. Zhang, et al., AdaCost: Misclassification Cost-Sensitive Boosting, In: Sixteenth International Conference on Machine Learning, (1999), 97–105. |
| [12] | Y. Freund and R.E. Schapire, A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting, J. Comput. Syst. Sci., 55(1997), 119–139. |
| [13] | J. Long, E. Shelhamer and T. Darrell, Fully convolutional networks for semantic segmentation, In: IEEE Conference on Computer Vision and Pattern Recognition, 39(2015), 640–651. |
| [14] | T. J. Brazil, Causal-Convolution-A New Method for the Transient Analysis of Linear Systems at Microwave Frequencies, IEEE T. Microw. Theory, 43(1995), 315–323. |
| [15] | A. V. D. Oord, S. Dieleman and H. Zen, et al., WaveNet: A Generative Model for Raw Audio, preprint, arXiv:1609.03499. |
| [16] | K. He, X. Zhang and S. Ren, et al., Deep Residual Learning for Image Recognition, In: IEEE Conference on Computer Vision and Pattern Recognition, (2016), 770–778. |
| [17] | P. Branco, L. Torgo and R. Ribeiro, A survey of predictive modelling under imbalanced distributions. preprint, arXiv:1505.01658. |
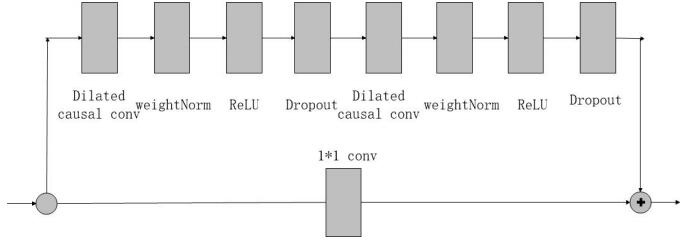
Figures(1) / Tables(5)

Hanyu Zhao, Chao Che, Bo Jin, Xiaopeng Wei. A viral protein identifying framework based on temporal convolutional network[J]. Mathematical Biosciences and Engineering, 2019, 16(3): 1709-1717. doi: 10.3934/mbe.2019081







 DownLoad:
DownLoad: