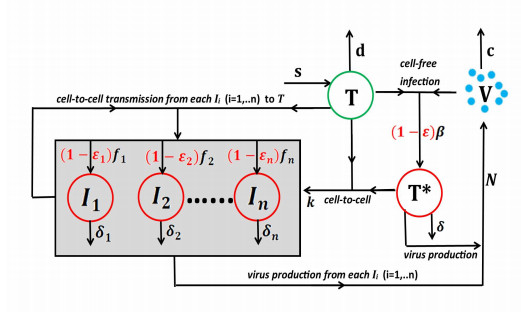
HIV infection remains a serious global public health problem. Although current drug treatment is effective and can reduce plasma viral loads below the level of detection, it cannot eradicate the virus. The reasons for the low virus persistence despite long-term therapy have not been fully elucidated. In addition, multiple HIV infection, i.e., infection of a cell by multiple viruses, is common and can facilitate viral recombination and mutations, evading the immune system and conferring resistance to drug treatment. The mechanisms for multiple HIV infection formation and their respective contributions remain unclear. To answer these questions, we developed a mathematical modeling framework that encompasses cell-free viral infection and cell-to-cell spread. We fit sub-models that only have one transmission route and the full model containing both to the multi-infection data from HIV-infected patients, and show that the multi-infection data can only be reproduced if these two transmission routes are both considered. Computer simulations with the best-fitting parameter values indicate that cell-to-cell spread leads to the majority of multiple infection and also accounts for the majority of overall infection. Sensitivity analysis shows that cell-to-cell spread has reduced susceptibility to treatment and may explain low HIV persistence. Taken together, this work indicates that cell-to-cell spread plays a crucial role in the development of HIV multi-infection and low HIV persistence despite long-term therapy, and therefore has important implications for understanding HIV pathogenesis and developing more effective treatment strategies to control or even eliminate the disease.
Citation: Sophia Y. Rong, Ting Guo, J. Tyler Smith, Xia Wang. The role of cell-to-cell transmission in HIV infection: insights from a mathematical modeling approach[J]. Mathematical Biosciences and Engineering, 2023, 20(7): 12093-12117. doi: 10.3934/mbe.2023538
HIV infection remains a serious global public health problem. Although current drug treatment is effective and can reduce plasma viral loads below the level of detection, it cannot eradicate the virus. The reasons for the low virus persistence despite long-term therapy have not been fully elucidated. In addition, multiple HIV infection, i.e., infection of a cell by multiple viruses, is common and can facilitate viral recombination and mutations, evading the immune system and conferring resistance to drug treatment. The mechanisms for multiple HIV infection formation and their respective contributions remain unclear. To answer these questions, we developed a mathematical modeling framework that encompasses cell-free viral infection and cell-to-cell spread. We fit sub-models that only have one transmission route and the full model containing both to the multi-infection data from HIV-infected patients, and show that the multi-infection data can only be reproduced if these two transmission routes are both considered. Computer simulations with the best-fitting parameter values indicate that cell-to-cell spread leads to the majority of multiple infection and also accounts for the majority of overall infection. Sensitivity analysis shows that cell-to-cell spread has reduced susceptibility to treatment and may explain low HIV persistence. Taken together, this work indicates that cell-to-cell spread plays a crucial role in the development of HIV multi-infection and low HIV persistence despite long-term therapy, and therefore has important implications for understanding HIV pathogenesis and developing more effective treatment strategies to control or even eliminate the disease.

| [1] | WHO, HIV/AIDS: key facts, 2023. Available from: https://www.who.int/news-room/fact-sheets/detail/hiv-aids. |
| [2] | FDA Approval of HIV medicines, AIDSinfo, 2018. Available from: https://aidsinfo.nih.gov/understanding-hiv-aids/infographics/25/fda-approval-of-hiv-medicines. |
| [3] |
G. Dornadula, H. Zhang, B. VanUitert, J. Stern, L. Livornese, M. J. Ingerman, et al., Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy, JAMA, 282 (1999), 1627–1632. https://doi.org/10.1001/jama.282.17.1627 doi: 10.1001/jama.282.17.1627

|
| [4] |
F. Maldarelli, Targeting viral reservoirs: ability of antiretroviral therapy to stop viral replication, Curr. Opin. HIV AIDS, 6 (2011), 49–56. https://doi.org/10.1097/COH.0b013e32834134ea doi: 10.1097/COH.0b013e32834134ea
|
| [5] |
S. Palmer, F. Maldarelli, A. Wiegand, B. Bernstein, G. J. Hanna, S. C. Brun, et al., Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy, Proc. Natl. Acad. Sci. USA, 105 (2008), 3879–3884. https://doi.org/10.1073/pnas.0800050105 doi: 10.1073/pnas.0800050105
|
| [6] |
L. Shan, R. F. Siliciano, From reactivation of latent HIV-1 to elimination of the latent reservoir: the presence of multiple barriers to viral eradication, BioEssays, 35 (2013), 544–552. https://doi.org/10.1002/bies.201200170 doi: 10.1002/bies.201200170
|
| [7] |
N. Ching, O. O. Yang, J. G. Deville, K. Nielsen-Saines, B. J. Ank, M. S. Sim, et al., Pediatric HIV-1-specific cytotoxic T-lymphocyte responses suggesting ongoing viral replication despite combination antiretroviral therapy, Pediatr. Res., 61 (2007), 692–697. https://doi.org/10.1203/pdr.0b013e31805365ef doi: 10.1203/pdr.0b013e31805365ef
|
| [8] |
T. Guo, Z. Qiu, L. Rong, Modeling the role of macrophages in HIV persistence during antiretroviral therapy, J. Math. Biol., 81 (2020), 369–402. https://doi.org/10.1007/s00285-020-01513-x doi: 10.1007/s00285-020-01513-x
|
| [9] |
J. Zhang, A. S. Perelson, Contribution of follicular dendritic cells to persistent HIV viremia, J. Virol., 87 (2013), 7893–7901. https://doi.org/10.1128/JVI.00556-13 doi: 10.1128/JVI.00556-13
|
| [10] |
J. Feldmann, O. Schwartz, HIV-1 virological synapse: live imaging of transmission, Viruses, 2 (2010), 1666–1680. https://doi.org/10.3390/v2081666 doi: 10.3390/v2081666
|
| [11] |
Q. J. Sattentau, Cell-to-cell spread of retroviruses, Viruses, 2 (2010), 1306–1321. https://doi.org/10.3390/v2061306 doi: 10.3390/v2061306
|
| [12] |
W. Hübner, G. McNerney, P. Chen, B. M. Dale, R. E. Gordon, F. Y. S. Chuang, et al., Quantitative 3D video microscopy of HIV transfer across T cell virological synapses, Science, 323 (2009), 1743–1747. https://doi.org/10.1126/science.1167525 doi: 10.1126/science.1167525
|
| [13] |
L. M. Agosto, P. Uchil, W. Mothes, HIV cell-to-cell transmission: effects on pathogenesis and antiretroviral therapy, Trends Microbiol., 23 (2015), 289–295. https://doi.org/10.1016/j.tim.2015.02.003 doi: 10.1016/j.tim.2015.02.003
|
| [14] |
P. Zhong, L. M. Agosto, J. B. Munro, W. Mothes, Cell-to-cell transmission of viruses, Curr. Opin. Virol., 3 (2013), 44–50. https://doi.org/10.1016/j.coviro.2012.11.004 doi: 10.1016/j.coviro.2012.11.004
|
| [15] |
A. Sigal, J. T. Kim, A. B. Balazs, E. Dekel, A. Mayo, R. Milo, et al., Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy, Nature, 477 (2011), 95–98. https://doi.org/10.1038/nature10347 doi: 10.1038/nature10347
|
| [16] |
N. L. Komarova, D. Wodar, Virus dynamics in the presence of synaptic transmission, Math. Biosci., 242 (2013), 161–171. https://doi.org/10.1016/j.mbs.2013.01.003 doi: 10.1016/j.mbs.2013.01.003
|
| [17] |
A. Jung, R. Maier, J. P. Vartanian, G. Bocharov, V. Jung, U. Fischer, et al., Recombination: multiply infected spleen cells in HIV patients, Nature, 418 (2002), 144. https://doi.org/10.1038/418144a doi: 10.1038/418144a
|
| [18] |
J. J. Mattapallil, D. C. Douek, B. Hill, Y. Nishimura, M. Martin, M. Roederer, Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection, Nature, 434 (2005), 1093–1097. https://doi.org/10.1038/nature03501 doi: 10.1038/nature03501
|
| [19] |
A. Rebecca, M. Nicola, M. Ivonne, E. Jones, Q. J. Sattentau, Multiple proviral integration events after virological synapse-mediated HIV-1 spread, Virology, 443 (2013), 143–149. https://doi.org/10.1016/j.virol.2013.05.005 doi: 10.1016/j.virol.2013.05.005
|
| [20] |
N. M. Dixit, A. S. Perelson, Multiplicity of human immunodeficiency virus infections in lymphoid tissue, J. Virol., 78 (2004), 8942–8945. https://doi.org/10.1128/JVI.78.16.8942-8945.2004 doi: 10.1128/JVI.78.16.8942-8945.2004
|
| [21] |
N. M. Dixit, A. S. Perelson, HIV dynamics with multiple infections of target cells, Proc. Natl. Acad. Sci. USA, 102 (2005), 8198–8203. https://doi.org/10.1073/pnas.0407498102 doi: 10.1073/pnas.0407498102
|
| [22] |
Y. Ito, A. Tauzin, A. Remion, K. Ejima, F. Mammano, S. Iwami, Dynamics of HIV-1 coinfection in different susceptible target cell populations during cell-free infection, J. Theor. Biol., 455 (2018), 39–46. https://doi.org/10.1016/j.jtbi.2018.06.025 doi: 10.1016/j.jtbi.2018.06.025
|
| [23] |
X. Wang, L. Rong, HIV low viral load persistence under treatment: insights from a model of cell-to-cell viral transmission, Appl. Math. Lett., 94 (2019), 44–51. https://doi.org/10.1016/j.aml.2019.02.019 doi: 10.1016/j.aml.2019.02.019
|
| [24] |
A. S. Perelson, P. W. Nelson, Mathematical analysis of HIV-1 dynamics in vivo, SIAM Rev., 41 (1999), 3–44. https://doi.org/10.1137/S0036144598335107 doi: 10.1137/S0036144598335107
|
| [25] | M. A. Nowak, R. M. May, Virus Dynamics: Mathematical Principles of Immunology and Virology, Oxfors: Oxford University Press, 2000. |
| [26] |
H. Mohri, S. Bonhoeffer, S. Monard, A. S. Perelson, D. D. Ho, Rapid turnover of T lymphocytes in SIV-infected rhesus macaques, Science, 279 (1998), 1223–1227. https://doi.org/10.1126/science.279.5354.1223 doi: 10.1126/science.279.5354.1223
|
| [27] | L. Rong, A. Perelson, Modeling HIV persistence, the latent reservoir, and viral blips, J. Theor. Biol., 260 (2009), 308–331. https://doi.org/10.1016/j.jtbi.2009.06.011 |
| [28] |
T. Guo, Z. Qiu, The effects of CTL immune response on HIV infection model with potent therapy, latently infected cells and cell-to-cell viral transmission, Math. Biosci. Eng., 16 (2019), 6822–6841. https://doi.org/10.3934/mbe.2019341 doi: 10.3934/mbe.2019341
|
| [29] |
X. Wang, S. Tang, X. Song, L. Rong, Mathematical analysis of an HIV latent infection model including both virus-to-cell infection and cell-to-cell transmission, J. Biol. Dyn., 11 (2017), 455–483. https://doi.org/10.1080/17513758.2016.1242784 doi: 10.1080/17513758.2016.1242784
|
| [30] |
P. V. den Driessche, J. Watmough, Reproduction numbers and sub-threshold endemic equilibria for compartmental models of disease transmission, Math. Biosci., 180 (2002), 29–48. https://doi.org/10.1016/S0025-5564(02)00108-6 doi: 10.1016/S0025-5564(02)00108-6
|
| [31] |
M. Bofill, G. Janossy, C. A. Lee, D. Macdonald-Burns, A. N. Phillips, C. Sabin, et al., Laboratory control values for CD4 and CD8 T lymphocytes. Implications for HIV-1 diagnosis, Clin. Exp. Immunol., 88 (1992), 243–252. https://doi.org/10.1111/j.1365-2249.1992.tb03068.x doi: 10.1111/j.1365-2249.1992.tb03068.x
|
| [32] |
N. L. Komarova, D. N. Levy, D. Wodarz, Effect of synaptic transmission on viral fitness in HIV infection, PLoS One, 7 (2012), e48361. https://doi.org/10.1371/journal.pone.0048361 doi: 10.1371/journal.pone.0048361
|
| [33] |
N. Komarova, D. Levy, D. Wodarz, Synaptic transmission and the susceptibility of HIV infection to anti-viral drugs, Sci. Rep., 3 (2013), 2103. https://doi.org/10.1038/srep02103 doi: 10.1038/srep02103
|
| [34] |
M. Boullé, T. Müller, S. Dähling, Y. Ganga, L. Jackson, D. Mahamed, et al., HIV cell-to-cell spread results in earlier onset of viral gene expression by multiple infections per cell, PLoS Pathog., 12 (2016), e1005964. https://doi.org/10.1371/journal.ppat.1005964 doi: 10.1371/journal.ppat.1005964
|
| [35] |
A. R. Templeton, M. G. Kramer, J. Jarvis, J. Kowalski, S. Gange, M. F. Schneider, et al., Multiple-infection and recombination in HIV-1 within a longitudinal cohort of women, Retrovirology, 6 (2009), 54. https://doi.org/10.1186/1742-4690-6-54 doi: 10.1186/1742-4690-6-54
|
| [36] |
J. Lama, The physiological relevance of CD4 receptor down-modulation during HIV infection, Curr. HIV Res., 1 (2003), 167–184. https://doi.org/10.2174/1570162033485276 doi: 10.2174/1570162033485276
|
| [37] |
K. Levesque, A. Finzi, J. Binette, E. A. Cohen, Role of CD4 receptor down-regulation during HIV-1 infection, Curr. HIV Res., 2 (2004), 51–59. https://doi.org/10.2174/1570162043485086 doi: 10.2174/1570162043485086
|
| [38] |
M. Nethe, B. Berkhout, A. C. van der Kuyl, Retroviral superinfection resistance, Retrovirology, 2 (2005), 52. https://doi.org/10.1186/1742-4690-2-52 doi: 10.1186/1742-4690-2-52
|
| [39] |
D. Wodarz, D. N. Levy, Effect of different modes of viral spread on the dynamics of multiply infected cells in human immunodeficiency virus infection, J. R. Soc. Interface, 8 (2011), 289–300. https://doi.org/10.1098/rsif.2010.0266 doi: 10.1098/rsif.2010.0266
|
| [40] |
T. Guo, Z. Qiu, K. Kitagawa, S. Iwami, L. Rong, Modeling HIV multiple infection, J. Theor. Biol., 509 (2021), 110502. https://doi.org/10.1016/j.jtbi.2020.110502 doi: 10.1016/j.jtbi.2020.110502
|
| [41] |
D. N. Levy, G. M. Aldrovandi, O. Kutsch, G. M. Shaw, Dynamics of HIV-1 recombination in its natural target cells, Proc. Natl. Acad. Sci. USA, 101 (2004), 4204–4209. https://doi.org/10.1073/pnas.0306764101 doi: 10.1073/pnas.0306764101
|
| [42] |
L. Josefsson, M. King, B. Makitalo, J. Brännström, W. Shao, F. Maldarelli, et al., Majority of CD4+ T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule, Proc. Natl. Acad. Sci. USA, 108 (2011), 11199–11204. https://doi.org/10.1073/pnas.1107729108 doi: 10.1073/pnas.1107729108
|
| [43] |
H. Song, E. E. Giorgi, V. V. Ganusov, F. Cai, G. Athreya, H. Yoon, et al., Tracking HIV-1 recombination to resolve its contribution to HIV-1 evolution in natural infection, Nat. Commun., 9 (2018), 1928. https://doi.org/10.1038/s41467-018-04217-5 doi: 10.1038/s41467-018-04217-5
|
| [44] |
A. Carvajal-Rodríguez, K. A. Crandall, D. Posada, Recombination favors the evolution of drug resistance in HIV-1 during antiretroviral therapy, Infect. Genet. Evol., 7 (2007), 476–483. https://doi.org/10.1016/j.meegid.2007.02.001 doi: 10.1016/j.meegid.2007.02.001
|
| [45] |
J. Rawson, O. A. Nikolaitchik, B. F. Keele, V. K. Pathak, Recombination is required for efficient HIV-1 replication and the maintenance of viral genome integrity, Nucleic Acids Res., 46 (2018), 10535–10545. https://doi.org/10.1093/nar/gky910 doi: 10.1093/nar/gky910
|
| [46] |
J. Kreger, N. L. Komarova, D. Wodarz, Effect of synaptic cell-to-cell transmission and recombination on the evolution of double mutants in HIV, J. R. Soc. Interface, 17 (2020), 20190832. https://doi.org/10.1098/rsif.2019.0832 doi: 10.1098/rsif.2019.0832
|
| [47] |
M. Arenas, N. M. Araujo, C. Branco, N. Castelhano, E. Castro-Nallar, M. Pérez-Losada, Mutation and recombination in pathogen evolution: relevance, methods and controversies, Infect. Genet. Evol., 63 (2018), 295–306. https://doi.org/10.1016/j.meegid.2017.09.029 doi: 10.1016/j.meegid.2017.09.029
|
| [48] |
M. Kearney, J. Spindler, W. Shao, S. Yu, E. M. Anderson, A. O'Shea, et al., Lack of detectable HIV-1 molecular evolution during suppressive antiretroviral therapy, PLoS Pathog., 10 (2014), e1004010. https://doi.org/10.1371/journal.ppat.1004010 doi: 10.1371/journal.ppat.1004010
|
| [49] |
G. Bozzi, F. R. Simonetti, S. A. Watters, E. M. Anderson, M. Gouzoulis, M. F. Kearney, et al., No evidence of ongoing HIV replication or compartmentalization in tissues during combination antiretroviral therapy: implications for HIV eradication, Sci. Adv., 5 (2019), eaav2045. https://doi.org/10.1126/sciadv.aav2045 doi: 10.1126/sciadv.aav2045
|





Figures(6) / Tables(3)

Sophia Y. Rong, Ting Guo, J. Tyler Smith, Xia Wang. The role of cell-to-cell transmission in HIV infection: insights from a mathematical modeling approach[J]. Mathematical Biosciences and Engineering, 2023, 20(7): 12093-12117. doi: 10.3934/mbe.2023538







 DownLoad:
DownLoad: