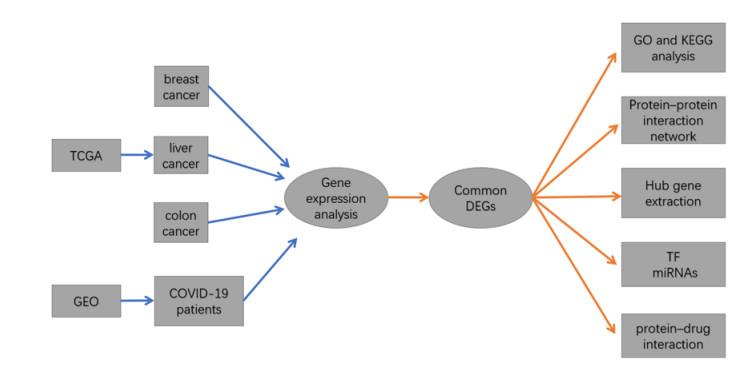
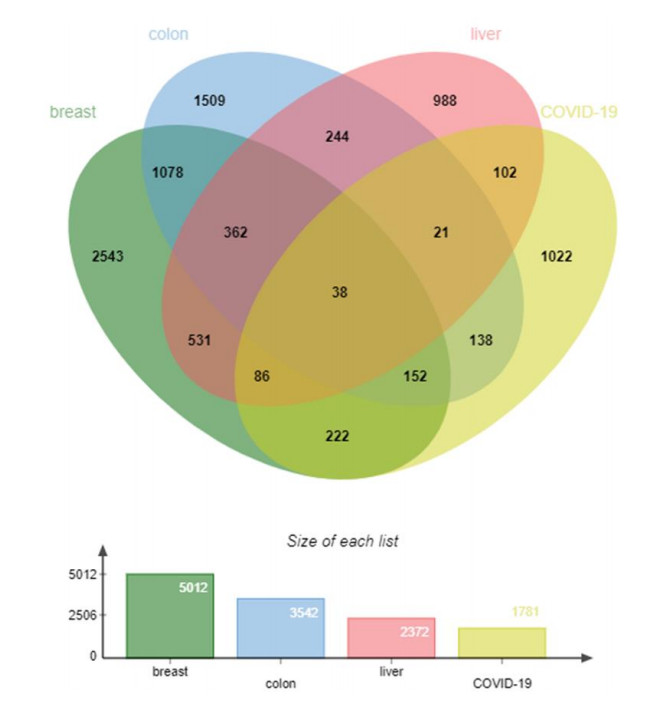
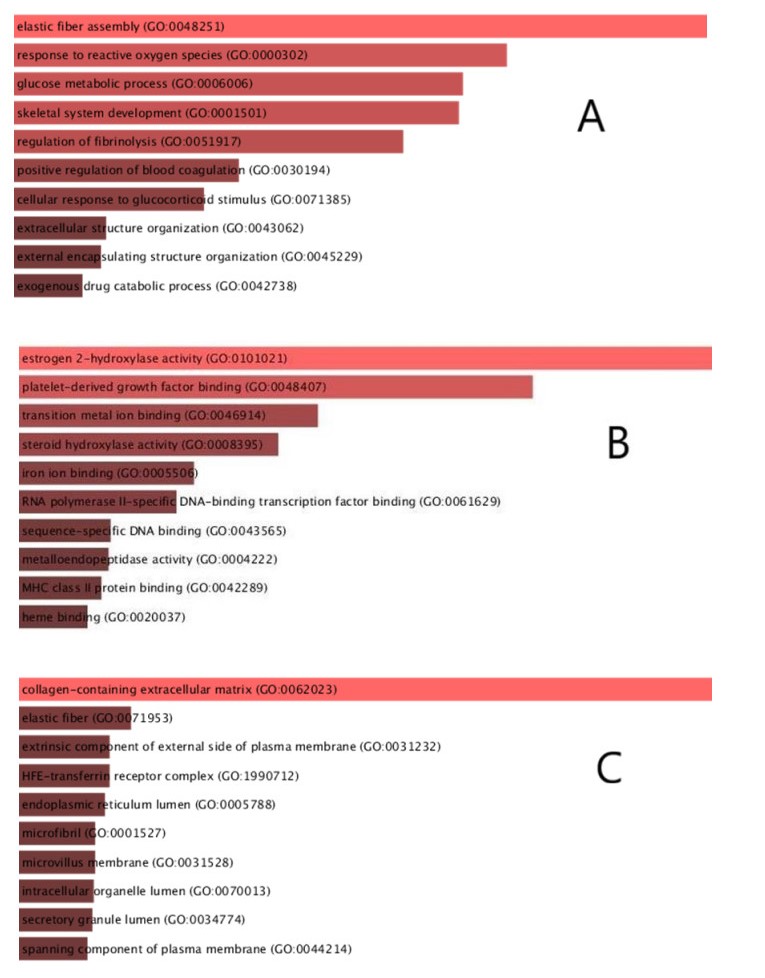
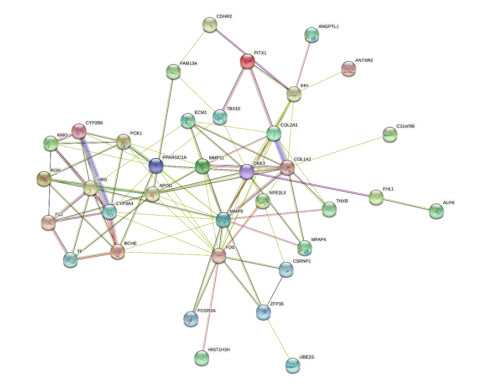
Severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), also known as COVID-19, is currently prevalent worldwide and poses a significant threat to human health. Individuals with cancer may have an elevated risk for SARS-CoV-2 infections and adverse outcomes. Therefore, it is necessary to explore the internal relationship between these two diseases. In this study, transcriptome analyses were performed to detect mutual pathways and molecular biomarkers in three types of common cancers of the breast, liver, colon, and COVID-19. Such analyses could offer a valuable understanding of the association between COVID-19 and cancer patients. In an analysis of RNA sequencing datasets for three types of cancers and COVID-19, we identified a sum of 38 common differentially expressed genes (DEGs). A variety of combinational statistical approaches and bioinformatics techniques were utilized to generate the protein-protein interaction (PPI) network. Subsequently, hub genes and critical modules were found using this network. In addition, a functional analysis was conducted using ontologies keywords, and pathway analysis was also performed. Some common associations between cancer and the risk and prognosis of COVID-19 were discovered. The datasets also revealed transcriptional factors-gene interplay, protein-drug interaction, and a DEGs-miRNAs coregulatory network with common DEGs. The potential medications discovered in this investigation could be useful in treating cancer and COVID-19.
Citation: Qinyan shen, Jiang wang, Liangying zhao. To investigate the internal association between SARS-CoV-2 infections and cancer through bioinformatics[J]. Mathematical Biosciences and Engineering, 2022, 19(11): 11172-11194. doi: 10.3934/mbe.2022521
Severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), also known as COVID-19, is currently prevalent worldwide and poses a significant threat to human health. Individuals with cancer may have an elevated risk for SARS-CoV-2 infections and adverse outcomes. Therefore, it is necessary to explore the internal relationship between these two diseases. In this study, transcriptome analyses were performed to detect mutual pathways and molecular biomarkers in three types of common cancers of the breast, liver, colon, and COVID-19. Such analyses could offer a valuable understanding of the association between COVID-19 and cancer patients. In an analysis of RNA sequencing datasets for three types of cancers and COVID-19, we identified a sum of 38 common differentially expressed genes (DEGs). A variety of combinational statistical approaches and bioinformatics techniques were utilized to generate the protein-protein interaction (PPI) network. Subsequently, hub genes and critical modules were found using this network. In addition, a functional analysis was conducted using ontologies keywords, and pathway analysis was also performed. Some common associations between cancer and the risk and prognosis of COVID-19 were discovered. The datasets also revealed transcriptional factors-gene interplay, protein-drug interaction, and a DEGs-miRNAs coregulatory network with common DEGs. The potential medications discovered in this investigation could be useful in treating cancer and COVID-19.

| [1] |
D. Cucinotta, M. Vanelli, WHO Declares COVID-19 a Pandemic, Acta Biomed., 91 (2020), 157–160. https://doi.org/10.23750/abm.v91i1.9397 doi: 10.23750/abm.v91i1.9397

|
| [2] | World Health Organization, Coronavirus disease (COVID-19). Available from: https://covid19.who.int/. |
| [3] |
K. A. Lee, W. Ma, D. R. Sikavi, D. A. Drew, L. H. Nguyen, R. Bowyer, et al., Cancer and risk of COVID-19 through a general community survey, Oncologist, 26 (2021), e182–e185. https://doi.org/10.1634/theoncologist.2020-0572 doi: 10.1634/theoncologist.2020-0572
|
| [4] |
W. Liang, W. Guan, R. Chen, W. Wang, J. Li, K. Xu, et al., Cancer patients in SARS-CoV-2 infection: a nationwide analysis in China, Lancet Oncol., 21 (2020), 335–337. https://doi.org/10.1016/S1470-2045(20)30096-6 doi: 10.1016/S1470-2045(20)30096-6
|
| [5] |
A. Lage, T. Crombet, Control of advanced cancer: the road to chronicity, Int. J. Environ. Res. Public Health, 8 (2011), 683–697. https://doi.org/10.3390/ijerph8030683 doi: 10.3390/ijerph8030683
|
| [6] |
R. Zheng, S. Zhang, H. Zeng, S. Wang, K. Sun, R. Chen, et al., Cancer incidence and mortality in China, 2016, J. Natl. Cancer Cent., 2 (2022), 1–9. https://doi.org/10.1016/j.jncc.2022.02.002 doi: 10.1016/j.jncc.2022.02.002
|
| [7] |
D. Blanco-Melo, B. E. Nilsson-Payant, W. C. Liu, S. Uhl, D. Hoagland, R. Moller, et al., Imbalanced host response to SARS-CoV-2 drives development of COVID-19, Cell, 181 (2020), 1036–1045. https://doi.org/10.1016/j.cell.2020.04.026 doi: 10.1016/j.cell.2020.04.026
|
| [8] |
P. Bardou, J. Mariette, F. Escudie, C. Djemiel, C. Klopp, Jvenn: an interactive Venn diagram viewer, BMC Bioinf., 15 (2014), 293. https://doi.org/10.1186/1471-2105-15-293 doi: 10.1186/1471-2105-15-293
|
| [9] |
A. Subramanian, P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, et al., Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles, Proc. Natl. Acad. Sci. USA, 102 (2005), 15545–15550. https://doi.org/10.1073/pnas.0506580102 doi: 10.1073/pnas.0506580102
|
| [10] |
M. V. Kuleshov, M. R. Jones, A. D. Rouillard, N. F. Fernandez, Q. Duan, Z. Wang, et al., Enrichr: a comprehensive gene set enrichment analysis web server 2016 update, Nucleic Acids Res., 44 (2016), W90–97. https://doi.org/10.1093/nar/gkw377 doi: 10.1093/nar/gkw377
|
| [11] |
C. H. Chin, S. H. Chen, H. H. Wu, C. W. Ho, M. T. Ko, C. Y. Lin, CytoHubba: Identifying hub objects and sub-networks from complex interactome, BMC Syst. Biol., 4 (2014), S11. https://doi.org/10.1186/1752-0509-8-S4-S11 doi: 10.1186/1752-0509-8-S4-S11
|
| [12] |
J. Xia, E. E. Gill, R. E. Hancock, NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data, Nat. Protoc., 10 (2015), 823–844. https://doi.org/10.1038/nprot.2015.052 doi: 10.1038/nprot.2015.052
|
| [13] |
A. Khan, O. Fornes, A. Stigliani, M. Gheorghe, J. A. Castro-Mondragon, R. van der Lee, et al., JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework, Nucleic Acids Res., 46 (2018), D260–D266. https://doi.org/10.1093/nar/gkx1126 doi: 10.1093/nar/gkx1126
|
| [14] |
P. Sethupathy, B. Corda, A. G. Hatzigeorgiou, TarBase: A comprehensive database of experimentally supported animal microRNA targets, RNA, 12 (2006), 192–197. https://doi.org/10.1261/rna.2239606 doi: 10.1261/rna.2239606
|
| [15] |
M. Yoo, J. Shin, J. Kim, K. A. Ryall, K. Lee, S. Lee, et al., DSigDB: drug signatures database for gene set analysis, Bioinformatics., 31 (2015), 3069–3071. https://doi.org/10.1093/bioinformatics/btv313 doi: 10.1093/bioinformatics/btv313
|
| [16] |
M. Giri, A. Puri, T. Wang, S. Guo, Comparison of clinical manifestations, pre-existing comorbidities, complications and treatment modalities in severe and non-severe COVID-19 patients: A systemic review and meta-analysis, Sci. Prog., 104 (2021), 368504211000906. https://doi.org/10.1177/00368504211000906 doi: 10.1177/00368504211000906
|
| [17] |
J. Yu, W. Ouyang, M. L. K. Chua, C. Xie, SARS-CoV-2 Transmission in Patients With Cancer at a Tertiary Care Hospital in Wuhan, China, JAMA Oncol., 6 (2020), 1108–1110. https://doi.org/10.1001/jamaoncol.2020.0980 doi: 10.1001/jamaoncol.2020.0980
|
| [18] |
J. Tian, X. Yuan, J. Xiao, Q. Zhong, C. Yang, B. Liu, et al., Clinical characteristics and risk factors associated with COVID-19 disease severity in patients with cancer in Wuhan, China: a multicentre, retrospective, cohort study, Lancet Oncol., 21 (2020), 893–903. https://doi.org/10.1016/S1470-2045(20)30309-0 doi: 10.1016/S1470-2045(20)30309-0
|
| [19] |
J. K. Goodrich, E. R. Davenport, A. G. Clark, R. E. Ley, The relationship between the human genome and microbiome comes into view, Annu. Rev. Genet., 51 (2017), 413–433. https://doi.org/10.1146/annurev-genet-110711-155532 doi: 10.1146/annurev-genet-110711-155532
|
| [20] |
D. Rajagopalan, S. Jha, An epi(c)genetic war: Pathogens, cancer and human genome, Biochim. Biophys. Acta Rev. Cancer, 1869 (2018), 333–345. https://doi.org/10.1016/j.bbcan.2018.04.003 doi: 10.1016/j.bbcan.2018.04.003
|
| [21] |
J. Minarovits, A. Demcsak, F. Banati, H. H. Niller, Epigenetic dysregulation in virus-associated neoplasms, Adv. Exp. Med. Biol., 879 (2016), 71–90. https://doi.org/10.1007/978-3-319-24738-0_4 doi: 10.1007/978-3-319-24738-0_4
|
| [22] |
V. J. Hofman, C. Moreilhon, P. D. Brest, S. Lassalle, K. Le Brigand, D. Sicard, et al., Gene expression profiling in human gastric mucosa infected with Helicobacter pylori, Mod. Pathol., 20 (2007), 974–989. https://doi.org/10.1038/modpathol.3800930 doi: 10.1038/modpathol.3800930
|
| [23] |
F. Geisslinger, A. M. Vollmar, K. Bartel, Cancer patients have a higher risk regarding COVID-19 - and vice versa?, Pharmaceuticals, 13 (2020), 143. https://doi.org/10.3390/ph13070143 doi: 10.3390/ph13070143
|
| [24] |
J. Zhang, H. Jiang, K. Du, T. Xie, B. Wang, C. Chen, et al., Pan-Cancer analysis of genomic and prognostic characteristics associated with coronavirus disease 2019 regulators, Front. Med., 8 (2021), 662460. https://doi.org/10.3389/fmed.2021.662460 doi: 10.3389/fmed.2021.662460
|
| [25] |
H. Goubran, J. Stakiw, J. Seghatchian, G. Ragab, T. Burnouf, SARS-CoV-2 and cancer: The intriguing and informative cross-talk, Transfus. Apher. Sci., (2022), 103488. https://doi.org/10.1016/j.transci.2022.103488 doi: 10.1016/j.transci.2022.103488
|
| [26] |
M. R. Rahman, T. Islam, T. Zaman, M. Shahjaman, M. R. Karim, F. Huq, et al., Identification of molecular signatures and pathways to identify novel therapeutic targets in Alzheimer's disease: Insights from a systems biomedicine perspective, Genomics, 112 (2020), 1290–1299. https://doi.org/10.1016/j.ygeno.2019.07.018 doi: 10.1016/j.ygeno.2019.07.018
|
| [27] |
Z. Nain, H. K. Rana, P. Lio, S. M. S. Islam, M. A. Summers, M. A. Moni, Pathogenetic profiling of COVID-19 and SARS-like viruses, Brief Bioinform., 22 (2021), 1175–1196. https://doi.org/10.1093/bib/bbaa173 doi: 10.1093/bib/bbaa173
|
| [28] |
T. Wang, Y. Zhang, J. Bai, Y. Xue, Q. Peng, MMP1 and MMP9 are potential prognostic biomarkers and targets for uveal melanoma, BMC Cancer, 21 (2021), 1068. https://doi.org/10.1186/s12885-021-08788-3 doi: 10.1186/s12885-021-08788-3
|
| [29] |
S. Mondal, N. Adhikari, S. Banerjee, S. A. Amin, T. Jha, Matrix metalloproteinase-9 (MMP-9) and its inhibitors in cancer: A minireview, Eur. J. Med. Chem., 194 (2020), 112260. https://doi.org/10.1016/j.ejmech.2020.112260 doi: 10.1016/j.ejmech.2020.112260
|
| [30] |
M. Bjorklund, E. Koivunen, Gelatinase-mediated migration and invasion of cancer cells, Biochim. Biophys. Acta, 1755 (2005), 37–69. https://doi.org/10.1016/j.bbcan.2005.03.001 doi: 10.1016/j.bbcan.2005.03.001
|
| [31] |
P. Chiranjeevi, K. M. Spurthi, N. S. Rani, G. R. Kumar, T. M. Aiyengar, M. Saraswati, et al., Gelatinase B (-1562C/T) polymorphism in tumor progression and invasion of breast cancer, Tumour Biol., 35 (2014), 1351–1356. https://doi.org/10.1007/s13277-013-1181-5 doi: 10.1007/s13277-013-1181-5
|
| [32] |
J. Niu, X. Gu, J. Turton, C. Meldrum, E. W. Howard, M. Agrez, Integrin-mediated signalling of gelatinase B secretion in colon cancer cells, Biochem. Biophys. Res. Commun., 249 (1998), 287–291. https://doi.org/10.1006/bbrc.1998.9128 doi: 10.1006/bbrc.1998.9128
|
| [33] |
S. Hazra, A. G. Chaudhuri, B. K. Tiwary, N. Chakrabarti, Matrix metallopeptidase 9 as a host protein target of chloroquine and melatonin for immunoregulation in COVID-19: A network-based meta-analysis, Life Sci., 257 (2020), 118096. https://doi.org/10.1016/j.lfs.2020.118096 doi: 10.1016/j.lfs.2020.118096
|
| [34] |
S. M. Sagar, F. R. Sharp, T. Curran, Expression of c-fos protein in brain: metabolic mapping at the cellular level, Science, 240 (1988), 1328–1331. https://doi.org/10.1126/science.3131879 doi: 10.1126/science.3131879
|
| [35] |
C. P. Matthews, N. H. Colburn, M. R. Young, AP-1 a target for cancer prevention, Curr. Cancer Drug Targets, 7 (2007), 317–324. https://doi.org/10.2174/156800907780809723 doi: 10.2174/156800907780809723
|
| [36] |
X. Qu, X. Yan, C. Kong, Y. Zhu, H. Li, D. Pan, et al., c-Myb promotes growth and metastasis of colorectal cancer through c-fos-induced epithelial-mesenchymal transition, Cancer Sci., 110 (2019), 3183–3196. https://doi.org/10.1111/cas.14141 doi: 10.1111/cas.14141
|
| [37] |
Y. Yu, D. Liu, Z. Liu, S. Li, Y. Ge, W. Sun, et al., The inhibitory effects of COL1A2 on colorectal cancer cell proliferation, migration, and invasion, J. Cancer, 9 (2018), 2953–2962. https://doi.org/10.7150/jca.25542 doi: 10.7150/jca.25542
|
| [38] |
C. Niehrs, Function and biological roles of the Dickkopf family of Wnt modulators, Oncogene, 25 (2006), 7469–7481. https://doi.org/10.1038/sj.onc.1210054 doi: 10.1038/sj.onc.1210054
|
| [39] |
J. Villar, H. Zhang, A. S. Slutsky, Lung repair and regeneration in ARDS: Role of PECAM1 and Wnt signaling, Chest, 155 (2019), 587–594. https://doi.org/10.1016/j.chest.2018.10.022 doi: 10.1016/j.chest.2018.10.022
|
| [40] |
E. Y. Choi, H. H. Park, H. Kim, H. N. Kim, I. Kim, S. Jeon, et al., Wnt5a and Wnt11 as acute respiratory distress syndrome biomarkers for severe acute respiratory syndrome coronavirus 2 patients, Eur. Respir. J., 56 (2020), 2001531. https://doi.org/10.1183/13993003.01531-2020 doi: 10.1183/13993003.01531-2020
|
| [41] |
Y. Zhang, C. Hu, WIF-1 and Ihh expression and clinical significance in patients with lung squamous cell carcinoma and adenocarcinoma, Appl. Immunohistochem. Mol. Morphol., 26 (2018), 454–461. https://doi.org/10.1097/PAI.0000000000000449 doi: 10.1097/PAI.0000000000000449
|
| [42] |
D. Tian, Z. Hu, CYP3A4-mediated pharmacokinetic interactions in cancer therapy, Curr. Drug Metab., 15(2014), 808–817. https://doi.org/10.2174/1389200216666150223152627 doi: 10.2174/1389200216666150223152627
|
| [43] |
Z. Nain, S. K. Barman, M. M. Sheam, S. B. Syed, A. Samad, J. Quinn, et al., Transcriptomic studies revealed pathophysiological impact of COVID-19 to predominant health conditions, Brief Bioinform., 22 (2021), bbab197. https://doi.org/10.1093/bib/bbab197 doi: 10.1093/bib/bbab197
|
| [44] |
H. L. Liu, I. J. Yeh, N. N. Phan, Y. H. Wu, M. C. Yen, J. H. Hung, et al., Gene signatures of SARS-CoV/SARS-CoV-2-infected ferret lungs in short- and long-term models, Infect. Genet. Evol., 85 (2020), 104438. https://doi.org/10.1016/j.meegid.2020.104438 doi: 10.1016/j.meegid.2020.104438
|
| [45] |
J. Zhao, H. Yu, Y. Liu, S. A. Gibson, Z. Yan, X. Xu, et al., Protective effect of suppressing STAT3 activity in LPS-induced acute lung injury, Am J Physiol Lung Cell Mol Physiol 311(2016), L868-L880. https://doi.org/10.1152/ajplung.00281.2016 doi: 10.1152/ajplung.00281.2016
|
| [46] |
E. K. Bajwa, P. C. Cremer, M. N. Gong, R. Zhai, L. Su, B. T. Thompson, et al., An NFKB1 promoter insertion/deletion polymorphism influences risk and outcome in acute respiratory distress syndrome among Caucasians, PLoS One, 6 (2011), e19469. https://doi.org/10.1371/journal.pone.0019469 doi: 10.1371/journal.pone.0019469
|
| [47] |
C. C. Sun, W. Zhu, S. J. Li, W. Hu, J. Zhang, Y. Zhuo, et al., FOXC1-mediated LINC00301 facilitates tumor progression and triggers an immune-suppressing microenvironment in non-small cell lung cancer by regulating the HIF1alpha pathway, Genome Med., 12 (2020), 77. https://doi.org/10.1186/s13073-020-00773-y doi: 10.1186/s13073-020-00773-y
|
| [48] |
J. Motalebzadeh, E. Eskandari, Transcription factors linked to the molecular signatures in the development of hepatocellular carcinoma on a cirrhotic background, Med. Oncol., 38 (2021), 121. https://doi.org/10.1007/s12032-021-01567-x doi: 10.1007/s12032-021-01567-x
|
| [49] |
M. Mahmoudian, E. Razmara, B. Mahmud Hussen, M. Simiyari, N. Lotfizadeh, H. Motaghed, et al., Identification of a six-microRNA signature as a potential diagnostic biomarker in breast cancer tissues, J. Clin. Lab. Anal., 35 (2021), e24010. https://doi.org/10.1002/jcla.24010 doi: 10.1002/jcla.24010
|
| [50] |
H. C. Li, Y. F. Chen, W. Feng, H. Cai, Y. Mei, Y. M. Jiang, et al., Loss of the Opa interacting protein 5 inhibits breast cancer proliferation through miR-139-5p/NOTCH1 pathway, Gene, 603 (2017), 1–8. https://doi.org/10.1016/j.gene.2016.11.046 doi: 10.1016/j.gene.2016.11.046
|
| [51] |
J. Tu, Z. Zhao, M. Xu, X. Lu, L. Chang, J. Ji, NEAT1 upregulates TGF-beta1 to induce hepatocellular carcinoma progression by sponging hsa-mir-139-5p, J. Cell Physiol., 233 (2018), 8578–8587. https://doi.org/10.1002/jcp.26524 doi: 10.1002/jcp.26524
|
| [52] |
D. Zhou, L. Dong, L. Yang, Q. Ma, F. Liu, Y. Li, et al., Identification and analysis of circRNA–miRNA–mRNA regulatory network in hepatocellular carcinoma, IET Syst. Biol., 14 (2020), 391–398. https://doi.org/10.1049/iet-syb.2020.0061 doi: 10.1049/iet-syb.2020.0061
|
| [53] |
Y. Xie, J. Li, P. Li, N. Li, Y. Zhang, H. Binang, et al., RNA-Seq profiling of serum exosomal circular RNAs reveals Circ-PNN as a potential biomarker for human colorectal cancer, Front. Oncol., 10 (2020), 982. https://doi.org/10.3389/fonc.2020.00982 doi: 10.3389/fonc.2020.00982
|
| [54] |
P. Ulivi, M. Canale, A. Passardi, G. Marisi, M. Valgiusti, G. L. Frassineti, et al., Circulating plasma levels of miR-20b, miR-29b and miR-155 as predictors of bevacizumab efficacy in patients with metastatic colorectal cancer, Int. J. Mol. Sci., 19 (2018), 307. https://doi.org/10.3390/ijms19010307 doi: 10.3390/ijms19010307
|
| [55] |
D. S. Kutilin, Regulation of gene expression of cancer/testis antigens in colorectal cancer patients, Mol. Biol., 54 (2020), 580–595. https://doi.org/10.31857/S0026898420040096 doi: 10.31857/S0026898420040096
|
| [56] |
H. Ni, B. Su, L. Pan, X. Li, X. Zhu, X. Chen, Functional variants inPXRare associated with colorectal cancer susceptibility in Chinese populations, Cancer Epidemiol., 39 (2015), 972–977. https://doi.org/10.1016/j.canep.2015.10.029 doi: 10.1016/j.canep.2015.10.029
|
| [57] |
H. Motieghader, M. Kouhsar, A. Najafi, B. Sadeghi, A. Masoudi-Nejad, mRNA-miRNA bipartite network reconstruction to predict prognostic module biomarkers in colorectal cancer stage differentiation, Mol. Biosyst., 13 (2017), 2168–2180. https://doi.org/10.1039/c7mb00400a doi: 10.1039/c7mb00400a
|
| [58] |
Y. H. Wu, I. J. Yeh, N. N. Phan, M. C. Yen, J. H. Hung, C. C. Chiao, et al., Gene signatures and potential therapeutic targets of Middle East respiratory syndrome coronavirus (MERS-CoV)-infected human lung adenocarcinoma epithelial cells, J. Microbiol. Immunol. Infect., 54 (2021), 845–857. https://doi.org/10.1016/j.jmii.2021.03.007 doi: 10.1016/j.jmii.2021.03.007
|
| [59] |
C. Li, A. Wu, K. Song, J. Gao, E. Huang, Y. Bai, et al., Identifying putative causal links between microRNAs and severe COVID-19 using Mendelian Randomization, Cells, 10 (2021), 3504. https://doi.org/10.3390/cells10123504 doi: 10.3390/cells10123504
|
| [60] |
M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, et al., Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro, Cell Res., 30 (2020), 269–271. https://doi.org/10.1038/s41422-020-0282-0 doi: 10.1038/s41422-020-0282-0
|
| [61] |
J. Liu, R. Cao, M. Xu, X. Wang, H. Zhang, H. Hu, et al., Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro, Cell Discov., 6 (2020), 16. https://doi.org/10.1038/s41421-020-0156-0 doi: 10.1038/s41421-020-0156-0
|
| [62] |
P. Gautret, J. C. Lagier, P. Parola, V. T. Hoang, L. Meddeb, M. Mailhe, et al., Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial, Int. J. Antimicrob. Agents., 56 (2020), 105949. https://doi.org/10.1016/j.ijantimicag.2020.105949 doi: 10.1016/j.ijantimicag.2020.105949
|
| [63] |
Y. Guo, Y. Wang, L. Cao, P. Wang, J. Qing, Q. Zheng, et al., A conserved inhibitory mechanism of a lycorine derivative against enterovirus and hepatitis C virus, Antimicrob. Agents. Chemother., 60 (2016), 913–924. https://doi.org/10.1128/AAC.02274-15 doi: 10.1128/AAC.02274-15
|
| [64] |
J. J. Nair, J. van Staden, Antiplasmodial lycorane alkaloid principles of the plant family Amaryllidaceae, Planta. Med., 85 (2019), 637–647. https://doi.org/10.1055/a-0880-5414 doi: 10.1055/a-0880-5414
|
| [65] |
X. Ge, X. Meng, D. Fei, K. Kang, Q. Wang, M. Zhao, Lycorine attenuates lipopolysaccharide-induced acute lung injury through the HMGB1/TLRs/NF-kappaB pathway, Biotech, 10 (2020), 369. https://doi.org/10.1007/s13205-020-02364-5 doi: 10.1007/s13205-020-02364-5
|
| [66] |
X. Wang, J. Lu, S. Ge, Y. Hou, T. Hu, Y. Lv, et al., Astemizole as a drug to inhibit the effect of SARS-COV-2 in vitro, Microb. Pathog., 156 (2021), 104929. https://doi.org/10.1016/j.micpath.2021.104929 doi: 10.1016/j.micpath.2021.104929
|

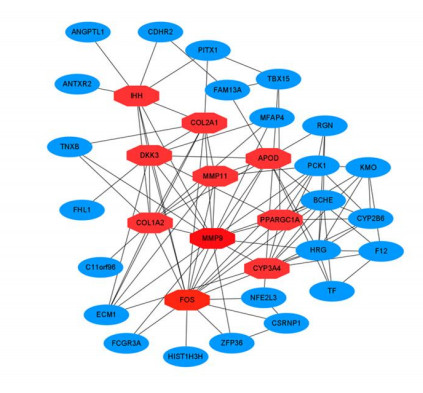
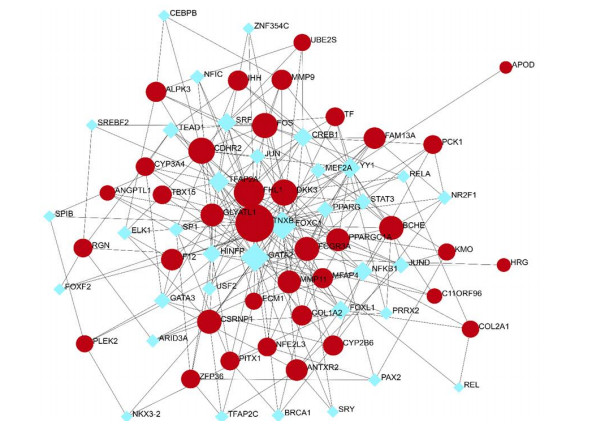
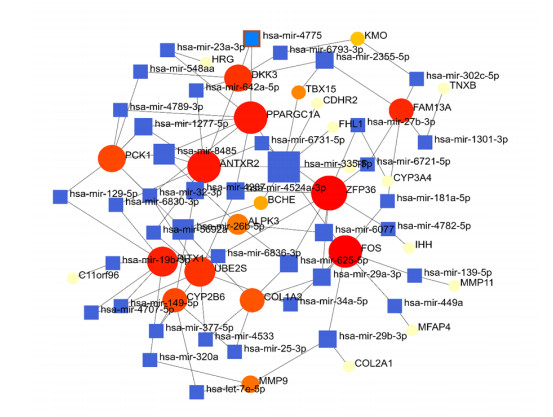
Figures(8) / Tables(4)

Qinyan shen, Jiang wang, Liangying zhao. To investigate the internal association between SARS-CoV-2 infections and cancer through bioinformatics[J]. Mathematical Biosciences and Engineering, 2022, 19(11): 11172-11194. doi: 10.3934/mbe.2022521







 DownLoad:
DownLoad: