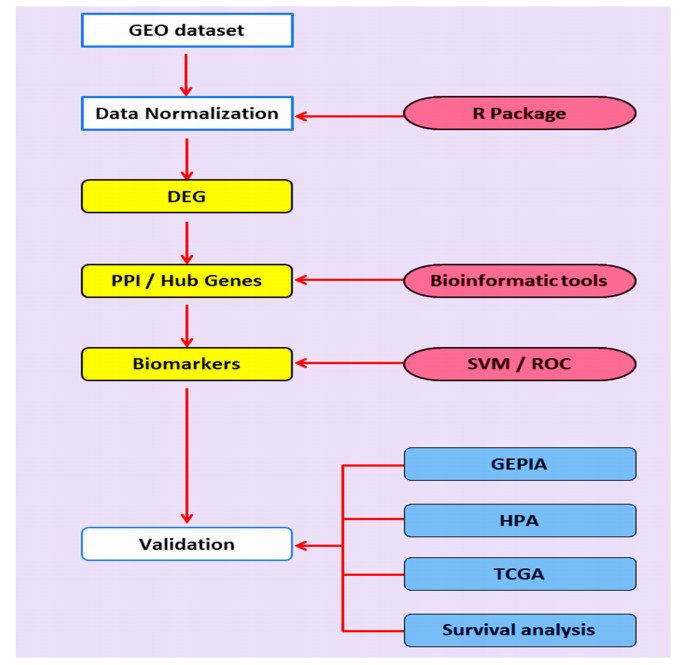
Colorectal cancer (CRC) is one of the most common malignancies worldwide. Biomarker discovery is critical to improve CRC diagnosis, however, machine learning offers a new platform to study the etiology of CRC for this purpose. Therefore, the current study aimed to perform an integrated bioinformatics and machine learning analyses to explore novel biomarkers for CRC prognosis. In this study, we acquired gene expression microarray data from Gene Expression Omnibus (GEO) database. The microarray expressions GSE103512 dataset was downloaded and integrated. Subsequently, differentially expressed genes (DEGs) were identified and functionally analyzed via Gene Ontology (GO) and Kyoto Enrichment of Genes and Genomes (KEGG). Furthermore, protein protein interaction (PPI) network analysis was conducted using the STRING database and Cytoscape software to identify hub genes; however, the hub genes were subjected to Support Vector Machine (SVM), Receiver operating characteristic curve (ROC) and survival analyses to explore their diagnostic values. Meanwhile, TCGA transcriptomics data in Gene Expression Profiling Interactive Analysis (GEPIA) database and the pathology data presented by in the human protein atlas (HPA) database were used to verify our transcriptomic analyses. A total of 105 DEGs were identified in this study. Functional enrichment analysis showed that these genes were significantly enriched in biological processes related to cancer progression. Thereafter, PPI network explored a total of 10 significant hub genes. The ROC curve was used to predict the potential application of biomarkers in CRC diagnosis, with an area under ROC curve (AUC) of these genes exceeding 0.92 suggesting that this risk classifier can discriminate between CRC patients and normal controls. Moreover, the prognostic values of these hub genes were confirmed by survival analyses using different CRC patient cohorts. Our results demonstrated that these 10 differentially expressed hub genes could be used as potential biomarkers for CRC diagnosis.
Citation: Ahmed Hammad, Mohamed Elshaer, Xiuwen Tang. Identification of potential biomarkers with colorectal cancer based on bioinformatics analysis and machine learning[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 8997-9015. doi: 10.3934/mbe.2021443
Colorectal cancer (CRC) is one of the most common malignancies worldwide. Biomarker discovery is critical to improve CRC diagnosis, however, machine learning offers a new platform to study the etiology of CRC for this purpose. Therefore, the current study aimed to perform an integrated bioinformatics and machine learning analyses to explore novel biomarkers for CRC prognosis. In this study, we acquired gene expression microarray data from Gene Expression Omnibus (GEO) database. The microarray expressions GSE103512 dataset was downloaded and integrated. Subsequently, differentially expressed genes (DEGs) were identified and functionally analyzed via Gene Ontology (GO) and Kyoto Enrichment of Genes and Genomes (KEGG). Furthermore, protein protein interaction (PPI) network analysis was conducted using the STRING database and Cytoscape software to identify hub genes; however, the hub genes were subjected to Support Vector Machine (SVM), Receiver operating characteristic curve (ROC) and survival analyses to explore their diagnostic values. Meanwhile, TCGA transcriptomics data in Gene Expression Profiling Interactive Analysis (GEPIA) database and the pathology data presented by in the human protein atlas (HPA) database were used to verify our transcriptomic analyses. A total of 105 DEGs were identified in this study. Functional enrichment analysis showed that these genes were significantly enriched in biological processes related to cancer progression. Thereafter, PPI network explored a total of 10 significant hub genes. The ROC curve was used to predict the potential application of biomarkers in CRC diagnosis, with an area under ROC curve (AUC) of these genes exceeding 0.92 suggesting that this risk classifier can discriminate between CRC patients and normal controls. Moreover, the prognostic values of these hub genes were confirmed by survival analyses using different CRC patient cohorts. Our results demonstrated that these 10 differentially expressed hub genes could be used as potential biomarkers for CRC diagnosis.

| [1] |
R. Siegel, D. Naishadham, A. Jemal, Cancer statistics, 2013, CA Cancer J. Clin., 63 (2013), 11-30. doi: 10.3322/caac.21166

|
| [2] | M. R. Sadeghi, F. Jeddi, N. Soozangar, M. H. Somi, N. Samadi, The role of Nrf2-Keap1 axis in colorectal cancer, progression, and chemoresistance, Tumor. Biol., 39 (2017), 1010428317705510. |
| [3] |
W. Chen, R. Zheng, P. D. Baade, S. Zhang, H. Zeng, F. Bray, et al., Cancer statistics in China, 2015, CA Cancer J. Clin., 66 (2016), 115-132. doi: 10.3322/caac.21338
|
| [4] | M. R. Sadeghi, F. Jeddi, N. Soozangar, M. H. Somi, N. Samadi, The role of Nrf2-Keap1 axis in colorectal cancer, progression, and chemoresistance, Tumour. Biol., 39 (2017), 1010428317705510. |
| [5] |
B. Raphael, R. Hruban, A. Aguirre, R. Moffitt, J. Yeh, C. Stewart, et al., Cancer Genome Atlas Research Network Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma, Cancer Cell, 32 (2017), 185-203. doi: 10.1016/j.ccell.2017.07.007
|
| [6] | I. Kinde, C. Bettegowda, Y. Wang, J. Wu, N. Agrawal, I. -M. Shih, et al., Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers, Sci. Transl. Med., 5 (2013), 167ra164-167ra164. |
| [7] |
M. Elshaer, A. I. ElManawy, A. Hammad, A. Namani, X. J. Wang, X. Tang, Integrated data analysis reveals significant associations of KEAP1 mutations with DNA methylation alterations in lung adenocarcinomas, Aging (Milano), 12 (2020), 7183-7206. doi: 10.18632/aging.103068
|
| [8] |
A. Hammad, Z. H. Zheng, A. Namani, M. Elshaer, X. J. Wang, X. Tang, Transcriptome analysis of potential candidate genes and molecular pathways in colitis-associated colorectal cancer of Mkp-1-deficient mice, BMC Cancer, 21 (2021), 607. doi: 10.1186/s12885-021-08200-0
|
| [9] |
B. Liang, C. Li, J. Zhao, Identification of key pathways and genes in colorectal cancer using bioinformatics analysis, Med. Oncol., 33 (2016), 016-0829. doi: 10.1007/s12032-015-0726-4
|
| [10] |
S. A. Bustin, S. Dorudi, Gene expression profiling for molecular staging and prognosis prediction in colorectal cancer, Expert Rev. Mol. Diagn., 4 (2004), 599-607. doi: 10.1586/14737159.4.5.599
|
| [11] |
V. Kulasingam, E. P. Diamandis, Strategies for discovering novel cancer biomarkers through utilization of emerging technologies, Nat. Clin. Pract. Oncol., 5 (2008), 588-599. doi: 10.1038/ncponc1187
|
| [12] |
M. Nannini, M. A. Pantaleo, A. Maleddu, A. Astolfi, S. Formica, G. Biasco, Gene expression profiling in colorectal cancer using microarray technologies: results and perspectives, Cancer Treat. Rev., 35 (2009), 201-209. doi: 10.1016/j.ctrv.2008.10.006
|
| [13] |
M. Ernst, T. L. Putoczki, Targeting IL-11 signaling in colon cancer, Oncotarget, 4 (2013), 1860. doi: 10.18632/oncotarget.1410
|
| [14] |
C. Isella, A. Terrasi, S. E. Bellomo, C. Petti, G. Galatola, A. Muratore, et al., Stromal contribution to the colorectal cancer transcriptome, Nat. Genet., 47 (2015), 312-319. doi: 10.1038/ng.3224
|
| [15] |
B. Vogelstein, N. Papadopoulos, V. E. Velculescu, S. Zhou, L. A. Diaz, K. W. Kinzler, Cancer genome landscapes, Science, 339 (2013), 1546-1558. doi: 10.1126/science.1235122
|
| [16] |
A. Hammad, Z. H. Zheng, Y. Gao, A. Namani, H. F. Shi, X. Tang, Identification of novel Nrf2 target genes as prognostic biomarkers in colitis-associated colorectal cancer in Nrf2-deficient mice, Life Sci., 238 (2019), 116968. doi: 10.1016/j.lfs.2019.116968
|
| [17] | K. GÜÇKIRAN, İ. Cantürk, L. ÖZYILMAZ, DNA microarray gene expression data classification using SVM, MLP, and RF with feature selection methods relief and LASSO, Süleyman Demirel Üniv. Fen Bilimleri Enst. Derg., 23 (2019), 126-132. |
| [18] |
N. S. Maurya, S. Kushwaha, A. Chawade, A. Mani, Transcriptome profiling by combined machine learning and statistical R analysis identifies TMEM236 as a potential novel diagnostic biomarker for colorectal cancer, Sci. Rep., 11 (2021), 14304. doi: 10.1038/s41598-021-92692-0
|
| [19] |
N. Auslander, A. B. Gussow, E. V. Koonin, Incorporating Machine Learning into Established Bioinformatics Frameworks, Int. J. Mol. Sci., 22 (2021), 2903. doi: 10.3390/ijms22062903
|
| [20] |
W. Lian, H. Jin, J. Cao, X. Zhang, T. Zhu, S. Zhao, et al., Identification of novel biomarkers affecting the metastasis of colorectal cancer through bioinformatics analysis and validation through qRT-PCR, Cancer Cell Int., 20 (2020), 105. doi: 10.1186/s12935-020-01180-4
|
| [21] |
L. Xu, R. Wang, J. Ziegelbauer, W. W. Wu, R. F. Shen, H. Juhl, et al., Transcriptome analysis of human colorectal cancer biopsies reveals extensive expression correlations among genes related to cell proliferation, lipid metabolism, immune response and collagen catabolism, Oncotarget, 8 (2017), 74703-74719. doi: 10.18632/oncotarget.20345
|
| [22] | J. Zhou, L. Li, L. Wang, X. Li, H. Xing, L. Cheng, Establishment of a SVM classifier to predict recurrence of ovarian cancer, Mol. Med. Rep., 18 (2018), 3589-3598. |
| [23] |
J. Mourao-Miranda, A. A. T. S. Reinders, V. Rocha-Rego, J. Lappin, J. Rondina, C. Morgan, et al., Individualized prediction of illness course at the first psychotic episode: a support vector machine MRI study, Psychol. Med., 42 (2012), 1037-1047. doi: 10.1017/S0033291711002005
|
| [24] |
X. Chen, Q. F. Wu, G. Y. Yan, RKNNMDA: Ranking-based KNN for MiRNA-Disease Association prediction, RNA Biol., 14 (2017), 952-962. doi: 10.1080/15476286.2017.1312226
|
| [25] | J. Zhi, J. Sun, Z. Wang, W. Ding, Support vector machine classifier for prediction of the metastasis of colorectal cancer, Int. J. Mol. Med., 41 (2018), 1419-1426. |
| [26] | M. N. Gabere, M. A. Hussein, M. A. Aziz, Filtered selection coupled with support vector machines generate a functionally relevant prediction model for colorectal cancer, Oncol. Targets Ther., 9 (2016), 3313-3325. |
| [27] |
Y. R. Liu, Y. Hu, Y. Zeng, Z. X. Li, H. B. Zhang, J. L. Deng, et al., Neurexophilin and PC-esterase domain family member 4 (NXPE4) and prostate androgen-regulated mucin-like protein 1 (PARM1) as prognostic biomarkers for colorectal cancer, J. Cell. Biochem., 120 (2019), 18041-18052. doi: 10.1002/jcb.29107
|
| [28] |
X. Song, T. Tang, C. Li, X. Liu, L. Zhou, CBX8 and CD96 Are Important Prognostic Biomarkers of Colorectal Cancer, Med. Sci. Monit., 24 (2018), 7820-7827. doi: 10.12659/MSM.908656
|
| [29] | R. C. Team, The R project for statistical computing Available at: https://www.r-project.org, Accessed January, 26 (2018). |
| [30] |
W. H. Da, B. T. Sherman, R. A. Lempicki, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources, Nat. Protoc., 4 (2009), 44-57. doi: 10.1038/nprot.2008.211
|
| [31] |
S. Friedman, P. H. Rubin, C. Bodian, E. Goldstein, N. Harpaz, D. H. Present, Screening and surveillance colonoscopy in chronic Crohns colitis, Gastroenterology, 120 (2001), 820-826. doi: 10.1053/gast.2001.22449
|
| [32] |
D. Szklarczyk, A. Franceschini, S. Wyder, K. Forslund, D. Heller, J. Huerta-Cepas, et al., STRING v10: protein-protein interaction networks, integrated over the tree of life, Nucleic Acids Res., 43 (2015), D447-D452. doi: 10.1093/nar/gku1003
|
| [33] |
P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, et al., Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Res., 13 (2003), 2498-2504. doi: 10.1101/gr.1239303
|
| [34] |
S. Babicki, D. Arndt, A. Marcu, Y. Liang, J. R. Grant, A. Maciejewski, et al., Heatmapper: web-enabled heat mapping for all, Nucleic Acids Res., 44 (2016), 17. doi: 10.1093/nar/gkv904
|
| [35] | J. Zhou, L. Li, L. Wang, X. Li, H. Xing, L. Cheng, Establishment of a SVM classifier to predict recurrence of ovarian cancer, Mol. Med. Rep., 18 (2018), 3589-3598. |
| [36] |
L. J. K. Wee, D. Simarmata, Y. W. Kam, L. F. P. Ng, J. C. Tong, SVM-based prediction of linear B-cell epitopes using Bayes Feature Extraction, BMC Genom., 11 (2010), S21. doi: 10.1186/1471-2164-11-S4-S21
|
| [37] |
Y. Hu, T. Hase, H. P. Li, S. Prabhakar, H. Kitano, S. K. Ng, et al., A machine learning approach for the identification of key markers involved in brain development from single-cell transcriptomic data, BMC Genom., 17 (2016), 1025-1025. doi: 10.1186/s12864-016-3317-7
|
| [38] |
C. D. A. Vanitha, D. Devaraj, M. Venkatesulu, Gene Expression Data Classification Using Support Vector Machine and Mutual Information-based Gene Selection, Proc. Comput. Sci., 47 (2015), 13-21. doi: 10.1016/j.procs.2015.03.178
|
| [39] |
N. S. Maurya, S. Kushwaha, A. Chawade, A. Mani, Transcriptome profiling by combined machine learning and statistical R analysis identifies TMEM236 as a potential novel diagnostic biomarker for colorectal cancer, Sci. Rep., 11 (2021), 021-92692. doi: 10.1038/s41598-020-79528-z
|
| [40] |
T. S. Furey, N. Cristianini, N. Duffy, D. W. Bednarski, M. Schummer, D. Haussler, Support vector machine classification and validation of cancer tissue samples using microarray expression data, Bioinformatics, 16 (2000), 906-914. doi: 10.1093/bioinformatics/16.10.906
|
| [41] | K. Liu, Q. Fu, Y. Liu, C. Wang, An integrative bioinformatics analysis of microarray data for identifying hub genes as diagnostic biomarkers of preeclampsia, Biosci. Rep., 39 (2019). |
| [42] |
L. K. Boroughs, R. J. DeBerardinis, Metabolic pathways promoting cancer cell survival and growth, Nat. Cell Biol., 17 (2015), 351-359. doi: 10.1038/ncb3124
|
| [43] |
Z. Tang, C. Li, B. Kang, G. Gao, C. Li, Z. Zhang, GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses, Nucleic Acids Res., 45 (2017), W98-W102. doi: 10.1093/nar/gkx247
|
| [44] |
I. M. Copple, The Keap1-Nrf2 cell defense pathway-a promising therapeutic target?, Adv. Pharmacol., 63 (2012), 43-79. doi: 10.1016/B978-0-12-398339-8.00002-1
|
| [45] |
K. Tong, O. Pellon-Cardenas, V. R. Sirihorachai, B. N. Warder, O. A. Kothari, A. O. Perekatt, et al., Degree of Tissue Differentiation Dictates Susceptibility to BRAF-Driven Colorectal Cancer, Cell Rep., 21 (2017), 3833-3845. doi: 10.1016/j.celrep.2017.11.104
|
| [46] |
F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre, A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J. Clin., 68 (2018), 394-424. doi: 10.3322/caac.21492
|
| [47] |
R. B. Sartor, Mechanisms of Disease: pathogenesis of Crohns disease and ulcerative colitis, Nat. Clin. Pract. Gastroenterol. Hepatol., 3 (2006), 390-407. doi: 10.1038/ncpgasthep0528
|
| [48] |
A. J. Schottelius, H. Dinter, Cytokines, NF-κB, Microenvironment, Intestinal Inflammation and Cancer, Cancer Treat. Res., 130 (2006), 67-87. doi: 10.1007/0-387-26283-0_3
|
| [49] |
C. Rubie, V. O. Frick, S. Pfeil, M. Wagner, O. Kollmar, B. Kopp, et al., Correlation of IL-8 with induction, progression and metastatic potential of colorectal cancer, World J. Gastroenterol., 13 (2007), 4996-5002. doi: 10.3748/wjg.v13.i37.4996
|
| [50] | B. Zhao, Z. Baloch, Y. Ma, Z. Wan, Y. Huo, F. Li, et al., Identification of Potential Key Genes and Pathways in Early-Onset Colorectal Cancer Through Bioinformatics Analysis, Cancer Control, 26 (2019), 1073274819831260. |
| [51] |
R. J. Wang, P. Wu, G. X. Cai, Z. M. Wang, Y. Xu, J. J. Peng, et al., Down-regulated MYH11 expression correlates with poor prognosis in stage Ⅱ and Ⅲ colorectal cancer, Asian Pac. J. Cancer Prev., 15 (2014), 7223-7228. doi: 10.7314/APJCP.2014.15.17.7223
|
| [52] |
N. Yamamoto, T. Oshima, K. Yoshihara, T. Aoyama, T. Hayashi, T. Yamada, et al., Clinicopathological significance and impact on outcomes of the gene expression levels of IGF-1, IGF-2 and IGF-1R, IGFBP-3 in patients with colorectal cancer: Overexpression of the IGFBP-3 gene is an effective predictor of outcomes in patients with colorectal cancer, Oncol. Lett., 13 (2017), 3958-3966. doi: 10.3892/ol.2017.5936
|
| [53] |
S. Wu, F. Wu, Z. Jiang, Identification of hub genes, key miRNAs and potential molecular mechanisms of colorectal cancer, Oncol. Rep., 38 (2017), 2043-2050. doi: 10.3892/or.2017.5930
|
| [54] |
T. Chen, J. Turner, S. McCarthy, M. Scaltriti, S. Bettuzzi, T. J. Yeatman, Clusterin-mediated apoptosis is regulated by adenomatous polyposis coli and is p21 dependent but p53 independent, Cancer Res., 64 (2004), 7412-7419. doi: 10.1158/0008-5472.CAN-04-2077
|
| [55] | W. Gomaa, M. Al-Ahwal, H. Al-Maghrabi, A. Buhmeida, M. Al-Qahtani, B. Al-Maghrabi, et al., Expression of clusterin in colorectal carcinoma in relation to clinicopathological criteria, Malays. J. Pathol., 39 (2017), 243-250. |
| [56] |
P. I. Artemaki, A. D. Sklirou, C. K. Kontos, A. A. Liosi, D. D. Gianniou, I. N. Papadopoulos, et al., High clusterin (CLU) mRNA expression levels in tumors of colorectal cancer patients predict a poor prognostic outcome, Clin. Biochem., 75 (2020), 62-69. doi: 10.1016/j.clinbiochem.2019.10.008
|
| [57] |
S. Mahner, C. Baasch, J. Schwarz, S. Hein, L. Wölber, F. Jänicke, et al., C-Fos expression is a molecular predictor of progression and survival in epithelial ovarian carcinoma, Br. J. Cancer, 99 (2008), 1269-1275. doi: 10.1038/sj.bjc.6604650
|
| [58] | R. Ashida, K. Tominaga, E. Sasaki, T. Watanabe, Y. Fujiwara, N. Oshitani, et al., AP-1 and colorectal cancer, Inflammopharmacology $V 13, (2006), 113-125. |
| [59] |
G. Chen, N. Han, G. Li, X. Li, Z. Li, Q. Li, Time course analysis based on gene expression profile and identification of target molecules for colorectal cancer, Cancer Cell Int., 16 (2016), 016-0296. doi: 10.1186/s12935-016-0291-8
|
| [60] |
X. Tan, M. Chen, MYLK and MYL9 expression in non-small cell lung cancer identified by bioinformatics analysis of public expression data, Tumor. Biol., 35 (2014), 12189-12200. doi: 10.1007/s13277-014-2527-3
|
| [61] |
B. Liang, C. Li, J. Zhao, Identification of key pathways and genes in colorectal cancer using bioinformatics analysis, Med. Oncol., 33 (2016), 111. doi: 10.1007/s12032-016-0829-6
|
| [62] |
G. Sun, Y. Li, Y. Peng, D. Lu, F. Zhang, X. Cui, et al., Identification of differentially expressed genes and biological characteristics of colorectal cancer by integrated bioinformatics analysis, J. Cell. Physiol., 234 (2019), 15215-15224. doi: 10.1002/jcp.28163
|
| [63] | J. E. Drew, A. J. Farquharson, C. D. Mayer, H. F. Vase, P. J. Coates, R. J. Steele, et al., Predictive gene signatures: molecular markers distinguishing colon adenomatous polyp and carcinoma, PLoS One, 9 (2014). |
| [64] | T. Yamane, K. Asanoma, H. Kobayashi, G. Liu, H. Yagi, T. Ohgami, et al., Identification of the Critical Site of Calponin 1 for Suppression of Ovarian Cancer Properties, Anticancer Res., 35 (2015), 5993-5999. |
| [65] |
Z. Y. Lin, W. L. Chuang, Genes responsible for the characteristics of primary cultured invasive phenotype hepatocellular carcinoma cells, Biomed. Pharmacother., 66 (2012), 454-458. doi: 10.1016/j.biopha.2012.04.001
|
| [66] | W. Xie, J. Zhang, P. Zhong, S. Qin, H. Zhang, X. Fan, et al., Expression and potential prognostic value of histone family gene signature in breast cancer, Exp. Ther. Med., 18 (2019), 4893-4903. |
| [67] |
V. Afshar-Kharghan, The role of the complement system in cancer, J. Clin. Invest., 127 (2017), 780-789. doi: 10.1172/JCI90962
|
| [68] | X. Chen, C. C. Yan, X. Zhang, Z. H. You, Long non-coding RNAs and complex diseases: from experimental results to computational models, Briefings Bioinf., 18 (2017), 558-576. |
| [69] |
X. Chen, D. Xie, Q. Zhao, Z. H. You, MicroRNAs and complex diseases: from experimental results to computational models, Briefings Bioinf., 20 (2019), 515-539. doi: 10.1093/bib/bbx130
|
| [70] | X. Chen, L. Wang, J. Qu, N. N. Guan, J. Q. Li, Predicting miRNA-disease association based on inductive matrix completion, Bioinformatics, 34 (2018), 4256-4265. |
| [71] | C. C. Wang, C. D. Han, Q. Zhao, X. Chen, Circular RNAs and complex diseases: from experimental results to computational models, Briefings Bioinfo., 2021. |
| [72] |
K. Strimbu, J. A. Tavel, What are biomarkers?, Curr. Opin. HIV AIDS, 5 (2010), 463-466. doi: 10.1097/COH.0b013e32833ed177
|
 mbe-18-06-443 -Supplementary.pdf mbe-18-06-443 -Supplementary.pdf |
 |






Figures(7)

Ahmed Hammad, Mohamed Elshaer, Xiuwen Tang. Identification of potential biomarkers with colorectal cancer based on bioinformatics analysis and machine learning[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 8997-9015. doi: 10.3934/mbe.2021443







 DownLoad:
DownLoad: