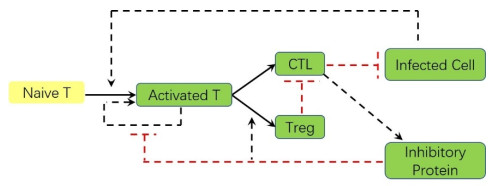
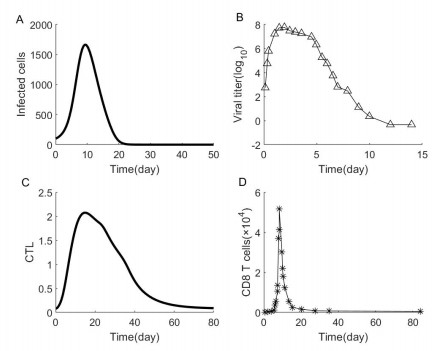
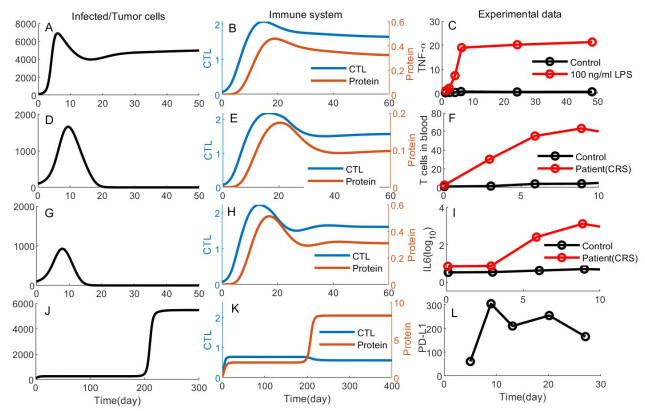
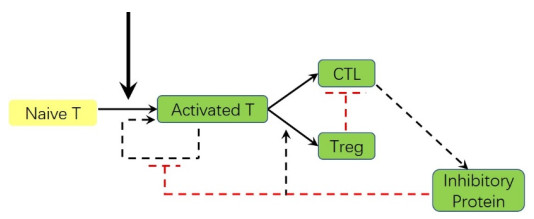
Adaptive immune responses can be activated by harmful stimuli. Upon activation, a cascade of biochemical events ensues the proliferation and the differentiation of T cells, which can remove the stimuli and undergo cell death to maintain immune cell homeostasis. However, normal immune processes can be disrupted by certain dysregulations, leading to pathological responses, such as cytokine storms and immune escape. In this paper, a qualitative mathematical model, composed of key feedback loops within the immune system, was developed to study the dynamics of various response behaviors. First, simulation results of the model well reproduce the results of several immune response processes, particularly pathological immune responses. Next, we demonstrated how the interaction of positive and negative feedback loops leads to irreversible bistable, reversible bistable and monostable, which characterize different immune response processes: cytokine storm, normal immune response, immune escape. The stability analyses suggest that the switch-like behavior is the basis of rapid activation of the immune system, and a balance between positive and negative regulation loops is necessary to prevent pathological responses. Furthermore, we have shown how the treatment moves the system back to a healthy state from the pathological immune response. The bistable mechanism that revealed in this work is helpful to understand the dynamics of different immune response processes.
Citation: Lingli Zhou, Fengqing Fu, Yao Wang, Ling Yang. Interlocked feedback loops balance the adaptive immune response[J]. Mathematical Biosciences and Engineering, 2022, 19(4): 4084-4100. doi: 10.3934/mbe.2022188
Adaptive immune responses can be activated by harmful stimuli. Upon activation, a cascade of biochemical events ensues the proliferation and the differentiation of T cells, which can remove the stimuli and undergo cell death to maintain immune cell homeostasis. However, normal immune processes can be disrupted by certain dysregulations, leading to pathological responses, such as cytokine storms and immune escape. In this paper, a qualitative mathematical model, composed of key feedback loops within the immune system, was developed to study the dynamics of various response behaviors. First, simulation results of the model well reproduce the results of several immune response processes, particularly pathological immune responses. Next, we demonstrated how the interaction of positive and negative feedback loops leads to irreversible bistable, reversible bistable and monostable, which characterize different immune response processes: cytokine storm, normal immune response, immune escape. The stability analyses suggest that the switch-like behavior is the basis of rapid activation of the immune system, and a balance between positive and negative regulation loops is necessary to prevent pathological responses. Furthermore, we have shown how the treatment moves the system back to a healthy state from the pathological immune response. The bistable mechanism that revealed in this work is helpful to understand the dynamics of different immune response processes.

| [1] |
C. A. Janeway, How the immune system works to protect the host from infection: A personal view, Proc. Natl. Acad. Sci., 98 (2001), 7461–7468. https://doi.org/10.1073/pnas.131202998 doi: 10.1073/pnas.131202998

|
| [2] |
R. D. Michalek, J. C. Rathmell, The metabolic life and times of a T‐cell, Immunol. Rev., 236 (2010), 190–202. https://doi.org/10.1111/j.1600-065X.2010.00911.x doi: 10.1111/j.1600-065X.2010.00911.x
|
| [3] |
K. C. Mccullough, A. Summerfield, Basic concepts of immune response and defense development, ILAR J., 46 (2005), 230–240. https://doi.org/10.1093/ilar.46.3.230 doi: 10.1093/ilar.46.3.230
|
| [4] |
A. Rahman, A. Tiwari, J. Narula, T. Hickling, Importance of feedback and feedforward loops to adaptive immune response modeling, CPT: Pharmacometrics Syst. Pharmacol., 7 (2018), 621–628. https://doi.org/10.1002/psp4.12352 doi: 10.1002/psp4.12352
|
| [5] |
C. C. Mok, C. S. Lau, Pathogenesis of systemic lupus erythematosus, J. Clin. Pathol., 56 (2003), 481–490. https://doi.org/10.1136/jcp.56.7.481 doi: 10.1136/jcp.56.7.481
|
| [6] |
A. Bhatia, Y. Kumar, Cellular and molecular mechanisms in cancer immune escape: a comprehensive review, Expert Rev. Clin. Immunol., 10 (2013), 41–62. https://doi.org/10.1586/1744666X.2014.865519 doi: 10.1586/1744666X.2014.865519
|
| [7] |
D. S. Vinay, E. P. Ryan, G. Pawelec, W. H. Tallib, J. Stagg, E. Elkord, et al., Immune evasion in cancer: Mechanistic basis and therapeutic strategies, Semin. Cancer Biol., 35 (2015), 185–198. https://doi.org/10.1016/j.semcancer.2015.03.004 doi: 10.1016/j.semcancer.2015.03.004
|
| [8] |
K. A. Abdel-Sater, Physiological positive feedback mechanisms, Am. J. Biomed. Sci., 3 (2011), 145–155. https://doi.org/10.5099/aj110200145 doi: 10.5099/aj110200145
|
| [9] |
N. Rapin, E. Mosekilde, O. Lund, Bistability in autoimmune diseases, Autoimmunity, 44 (2011), 256–260. https://doi.org/10.3109/08916934.2010.523233 doi: 10.3109/08916934.2010.523233
|
| [10] |
S. Wang, F. Xu F, L. Rong, Bistability analysis of an HIV model with immune response, J. Biol. Syst., 25 (2017), 677–695. https://doi.org/10.1142/S021833901740006X doi: 10.1142/S021833901740006X
|
| [11] |
C. Long, H. Qi, S. Huang, Mathematical modeling of cytotoxic lymphocyte-mediated immune response to hepatitis B virus infection, J. Biomed. Biotechnol., 2008 (2008), 743690. https://doi.org/10.1155/2008/743690 doi: 10.1155/2008/743690
|
| [12] |
K. León, A. Lage, J. Carneiro, Tolerance and immunity in a mathematical model of T-cell mediated suppression, J. Theor. Biol., 225 (2003), 107–126. https://doi.org/10.1016/S0022-5193(03)00226-1 doi: 10.1016/S0022-5193(03)00226-1
|
| [13] |
M. Robertson-Tessi, A. El-Kareh, A. Goriely, A mathematical model of tumor-immune interactions, J. Theor. Biol., 294 (2011), 56–73. https://doi.org/10.1016/j.jtbi.2011.10.027 doi: 10.1016/j.jtbi.2011.10.027
|
| [14] |
M. A. Vogelbaum, B. Otvos, B. Raychaudhuri, D. Hambardzumyan, J. Finke, J. Lathia, The role of Myeloid derived suppressor cells (MDSCs) in tumor-induced immunosuppression in human and murine gliomas, Neuro-Oncology, 16 (2014), 44–44. https://doi.org/10.1093/neuonc/nou209.10 doi: 10.1093/neuonc/nou209.10
|
| [15] |
X. Lai, A. Stiff, M. Duggan, R. Wesolowski, W. E. Carson Ⅲ, A. Friedman, Modeling combination therapy for breast cancer with BET and immune checkpoint inhibitors, Proc. Natl. Acad. Sci., 115 (2018), 5534–5539. https://doi.org/10.1073/pnas.1721559115 doi: 10.1073/pnas.1721559115
|
| [16] |
X. Lai, A. Friedman, How to schedule VEGF and PD-1 inhibitors in combination cancer therapy, BMC Syst. Biol., 13 (2019), 30. https://doi.org/10.1186/s12918-019-0706-y doi: 10.1186/s12918-019-0706-y
|
| [17] |
X. L. Lai, A. Friedman, Combination therapy for melanoma with BRAF/MEK inhibitor and immune checkpoint inhibitor: a mathematical model, BMC Syst. Biol., 11 (2017), 70. https://doi.org/10.1186/s12918-017-0446-9 doi: 10.1186/s12918-017-0446-9
|
| [18] |
A. Friedman, X. Lai, Combination therapy for cancer with oncolytic virus and checkpoint inhibitor: A mathematical model, PLoS ONE, 13 (2018), e0192449. https://doi.org/10.1371/journal.pone.0192449 doi: 10.1371/journal.pone.0192449
|
| [19] |
T. Kamada, Y. Togasjhi, C. Tay, D. Ha, A. Sasaki, Y. Nakamura, et al., PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer, Proc. Natl. Acad. Sci., 116 (2019), 9999–10008. https://doi.org/10.1073/pnas.1822001116 doi: 10.1073/pnas.1822001116
|
| [20] |
S. J. Rotz, D. Leino, S. Szabo, J. L. Mangino, B. K. Turpin, J. G. Pressey, Severe cytokine release syndrome in a patient receiving PD-1-directed therapy, Pediatr. Blood Cancer, 64 (2017), e26642. https://doi.org/10.1002/pbc.26642 doi: 10.1002/pbc.26642
|
| [21] |
N. L. Komarova, E. Barnes, P. Klenerman, D. Wodarz, Boosting immunity by antiviral drug therapy: A simple relationship among timing, efficacy, and success, Proc. Natl. Acad. Sci., , 100 (2003), 1855–1860. https://doi.org/10.1073/pnas.0337483100 doi: 10.1073/pnas.0337483100
|
| [22] |
N. L. Komarova, D. Wodarz, ODE models for oncolytic virus dynamics, J. Theor. Biol., 263 (2010), 530–543. https://doi.org/10.1016/j.jtbi.2010.01.009 doi: 10.1016/j.jtbi.2010.01.009
|
| [23] |
G. Mahlbacher, L. T. Curtis, J. Lowengrub, H. B. Frieboes, Mathematical modeling of tumor-associated macrophage interactions with the cancer microenvironment, J. Immunother. Cancer, 6 (2018), 10. https://doi.org/10.1186/s40425-017-0313-7 doi: 10.1186/s40425-017-0313-7
|
| [24] |
L. J. Carreno, P. A. González, A. M. Kalergis, Modulation of T cell function by TCR/pMHC binding kinetics, Immunobiology, 211 (2006), 47–64. https://doi.org/10.1016/j.imbio.2005.09.003 doi: 10.1016/j.imbio.2005.09.003
|
| [25] |
K. Li, R. William, Z. Yuan, C. Zhu, Single-molecule investigations of T-cell activation, Curr. Opin. Biomed. Eng., 12 (2019), 102–110. https://doi.org/10.1016/j.cobme.2019.10.005 doi: 10.1016/j.cobme.2019.10.005
|
| [26] |
P. Rozman, U. Svajger, The tolerogenic role of IFN-γ, Cytokine Growth Factor Rev., 41 (2018), 40–53. https://doi.org/10.1016/j.cytogfr.2018.04.001 doi: 10.1016/j.cytogfr.2018.04.001
|
| [27] |
J. D. Burke, H. A. Young, IFN-γ: A cytokine at the right time, is in the right place, Semin. Immunol., 43 (2019), 101280. https://doi.org/10.1016/j.smim.2019.05.002 doi: 10.1016/j.smim.2019.05.002
|
| [28] |
J. H. Esensten, Y. A. Helou, G. Chopra, A. Weiss, J. A. Bluestone, CD28 costimulation: from mechanism to therapy, Immunity, 44 (2016), 973–988. https://doi.org/10.1016/j.immuni.2016.04.020. doi: 10.1016/j.immuni.2016.04.020
|
| [29] |
M. Mandai, J. Hamamishi, K. Abiko, N. Matsumura, T. Baba, I. Konnishi, Dual faces of IFNγ in cancer progression: a role of PD-L1 induction in the determination of pro-and antitumor immunity, Clin. Cancer Res., 22 (2016), 2329–2334. https://doi.org/10.1158/1078-0432.CCR-16-0224 doi: 10.1158/1078-0432.CCR-16-0224
|
| [30] |
L. M. Francisco, V. H. Salinas, K. E. Brown, V. K. Vangugri, G. J. Freeman, V. K. Kuchroo, et al., PD-L1 regulates the development, maintenance, and function of induced regulatory T cells, J. Exp. Med., 206 (2009), 3015–3029. https://doi.org/10.1084/jem.20090847 doi: 10.1084/jem.20090847
|
| [31] |
E. Batlle, J. Massagué, Transforming growth factor-β signaling in immunity and cancer, Immunity, 50 (2019), 924–940. https://doi.org/10.1016/j.immuni.2019.03.024 doi: 10.1016/j.immuni.2019.03.024
|
| [32] |
P. Pandiyan, L. Zheng, S. Ishiharam, J. Reed, M. J. Lenardo, CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells, Nat. Immunol., 8 (2007), 1353–1362. https://doi.org/10.1038/ni1536 doi: 10.1038/ni1536
|
| [33] |
P. Trzonkowski, E. Szmit, J. Myliwska, A. Mysliwski, CD4+CD25+ T regulatory cells inhibit cytotoxic activity of CTL and NK cells in humans-impact of immunosenescence, Clin. Immunol., 119 (2006), 307–316. https://doi.org/10.1016/j.clim.2006.02.002 doi: 10.1016/j.clim.2006.02.002
|
| [34] |
L. Sewalt, K. Harley, P. V. Heijster, S. Balasuriya, Influences of Allee effects in the spreading of malignant tumours, J. Theor. Biol., 394 (2016), 77–92. https://doi.org/10.1016/j.jtbi.2015.12.024 doi: 10.1016/j.jtbi.2015.12.024
|
| [35] |
Y. V. Tyutyunov, S. Sen, L. I. Titova, M. Banerjee, Predator overcomes the Allee effect due to indirect prey-taxis, Ecol. Complexity, 39 (2019), 100772. https://doi.org/10.1016/j.ecocom.2019.100772 doi: 10.1016/j.ecocom.2019.100772
|
| [36] |
Y. Yu, J. J. Nieto, A. Torres, K. Wang, A viral infection model with a nonlinear infection rate, Boundary Value Probl., 2009 (2009), 958016. https://doi.org/10.1155/2009/958016 doi: 10.1155/2009/958016
|
| [37] |
S. Halle, O. Halle, R. Förster, Mechanisms and dynamics of T cell-mediated cytotoxicity in vivo, Trends Immunol., 38 (2017), 432–443. https://doi.org/10.1016/j.it.2017.04.002 doi: 10.1016/j.it.2017.04.002
|
| [38] |
B. Weigelin, M. Krause, P. Friedl, Cytotoxic T lymphocyte migration and effector function in the tumor microenvironment, Immunol. Lett., 138 (2011), 19–21. https://doi.org/10.1016/j.imlet.2011.02.016 doi: 10.1016/j.imlet.2011.02.016
|
| [39] |
J. R. Schoenborn, C. B. Wilson, Regulation of interferon-gamma during innate and adaptive immune responses, Adv. Immunol., 96 (2007), 41. https://doi.org/10.1016/S0065-2776(07)96002-2 doi: 10.1016/S0065-2776(07)96002-2
|
| [40] |
K. Abiko, N. Matsumura, J. Hamanishi, N. Horikawa, R. Murakami, K. Yamaguchi, et al., IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer, Br. J. Cancer, 112 (2015), 1501–1509. https://doi.org/10.1038/bjc.2015.101 doi: 10.1038/bjc.2015.101
|
| [41] |
H. Miao, J. A. Hollenbaugh, M. S. Zand, J. Holden-Wiltse, T. R. Mosmann, A. S. Perelson, et al., Quantifying the early immune response and adaptive immune response kinetics in mice infected with influenza A virus, J. Virol., 13 (2010), 6687–6698. https://doi.org/10.1128/JVI.00266-10 doi: 10.1128/JVI.00266-10
|
| [42] |
T. L. Hackett, R. Holloway, S. T. Holgate, J. A. Warner, Dynamics of pro-inflammatory and anti-inflammatory cytokine release during acute inflammation in chronic obstructive pulmonary disease: an ex vivo study, Respir Res., 9 (2008), 47. https://doi.org/10.1186/1465-9921-9-47 doi: 10.1186/1465-9921-9-47
|
| [43] |
K. A. Hay, L. A. Hanafi, D. Li, J. Gust, W. C. Liles, M. M. Wurfel, et al., Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy, Blood, 130 (2017), 2295–2306. https://doi.org/10.1182/blood-2017-06-793141 doi: 10.1182/blood-2017-06-793141
|
| [44] |
X. Jiang, J, Wang, X. Deng, F. Xiong, J. Ge, B. Xiang, et al., Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape, Mol. Cancer, 18 (2019), 10. https://doi.org/10.1186/s12943-018-0928-4. doi: 10.1186/s12943-018-0928-4
|
| [45] |
H. Dong, S. E. Strome, D. R. Salomao, H. Tamura, F. Hirano, D. B. Flies, et al., Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion, Nat. Med., 8 (2002), 793–800. https://doi.org/10.1038/nm730 doi: 10.1038/nm730
|
| [46] |
N. Patsoukis, J. Brown, V. Petkova, F. Liu, L. Li, V. A Boussiotis, Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation, Sci. Signaling, 5 (2012). https://doi.org/10.1126/scisignal.2002796 doi: 10.1126/scisignal.2002796
|
| [47] |
T. Noguchi, J. P. Ward, M. M. Gubin, C. D. Arthur, S. H. Lee, J. Hundal, et al., Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape, Cancer Immunol. Res., 5 (2017), 106–117. https://doi.org/10.1158/2326-6066.CIR-16-0391 doi: 10.1158/2326-6066.CIR-16-0391
|
Figures(8) / Tables(1)

Lingli Zhou, Fengqing Fu, Yao Wang, Ling Yang. Interlocked feedback loops balance the adaptive immune response[J]. Mathematical Biosciences and Engineering, 2022, 19(4): 4084-4100. doi: 10.3934/mbe.2022188







 DownLoad:
DownLoad: