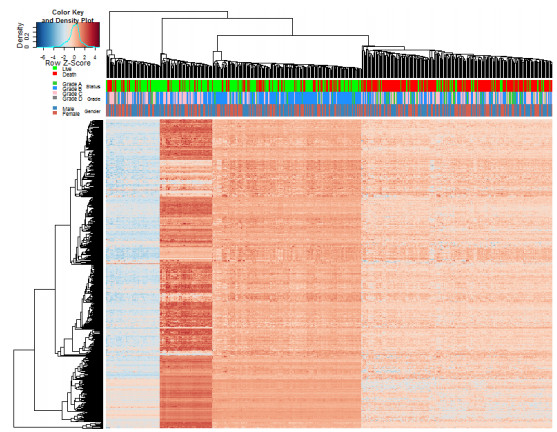
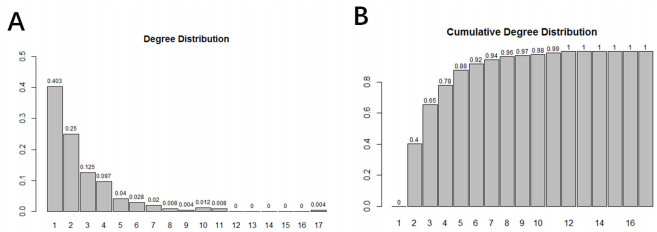
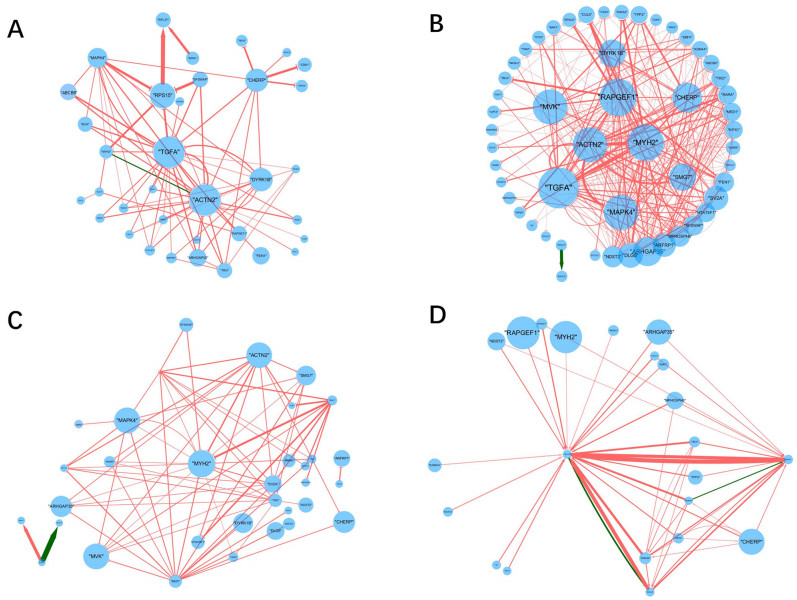
Inferring dynamic regulatory networks that rewire at different stages is a reasonable way to understand the mechanisms underlying cancer development. In this study, we reconstruct the stage-specific gene regulatory networks (GRNs) for colorectal cancer to understand dynamic changes of gene regulations along different disease stages. We combined multiple sets of clinical transcriptomic data of colorectal cancer patients and employed a supervised approach to select initial gene set for network construction. We then developed a dynamical system-based optimization method to infer dynamic GRNs by incorporating mutual information-based network sparsification and a dynamic cascade technique into an ordinary differential equations model. Dynamic GRNs at four different stages of colorectal cancer were reconstructed and analyzed. Several important genes were revealed based on the rewiring of the reconstructed GRNs. Our study demonstrated that reconstructing dynamic GRNs based on clinical transcriptomic profiling allows us to detect the dynamic trend of gene regulation as well as reveal critical genes for cancer development which may be important candidates of master regulators for further experimental test.
Citation: Ancheng Deng, Xiaoqiang Sun. Dynamic gene regulatory network reconstruction and analysis based on clinical transcriptomic data of colorectal cancer[J]. Mathematical Biosciences and Engineering, 2020, 17(4): 3224-3239. doi: 10.3934/mbe.2020183
Inferring dynamic regulatory networks that rewire at different stages is a reasonable way to understand the mechanisms underlying cancer development. In this study, we reconstruct the stage-specific gene regulatory networks (GRNs) for colorectal cancer to understand dynamic changes of gene regulations along different disease stages. We combined multiple sets of clinical transcriptomic data of colorectal cancer patients and employed a supervised approach to select initial gene set for network construction. We then developed a dynamical system-based optimization method to infer dynamic GRNs by incorporating mutual information-based network sparsification and a dynamic cascade technique into an ordinary differential equations model. Dynamic GRNs at four different stages of colorectal cancer were reconstructed and analyzed. Several important genes were revealed based on the rewiring of the reconstructed GRNs. Our study demonstrated that reconstructing dynamic GRNs based on clinical transcriptomic profiling allows us to detect the dynamic trend of gene regulation as well as reveal critical genes for cancer development which may be important candidates of master regulators for further experimental test.

| [1] |
X. Sun, B. Hu, Mathematical modeling and computational prediction of cancer drug resistance, Briefings Bioinf., 19 (2018), 1382-1399. doi: 10.1093/bib/bbx065

|
| [2] | B. H. Liu, Differential Coexpression Network Analysis for Gene Expression Data, in Computational Systems Biology: Methods and Protocols (ed T. Huang), Springer, New York, (2018), 155-165. |
| [3] |
K. Basso, A. A. Margolin, G. Stolovitzky, U. Klein, R. Dalla-Favera, A. Califano, Reverse engineering of regulatory networks in human B cells, Nat. Genet., 37 (2005), 382-390. doi: 10.1038/ng1532
|
| [4] | R. Tibshirani, Regression Shrinkage and Selection Via the Lasso, J. R. Stat. Soc., 58 (1996), 267-288. |
| [5] |
V. A. Huynh-Thu, A. Irrthum, L. Wehenkel, P. Geurts, Inferring Regulatory Networks from Expression Data Using Tree-Based Methods, PLOS ONE, 5 (2010), e12776. doi: 10.1371/journal.pone.0012776
|
| [6] |
N. Le Novère, Quantitative and logic modelling of molecular and gene networks, Nat. Rev. Genet., 16 (2015), 146-158. doi: 10.1038/nrg3885
|
| [7] | C. H. A. Higa, T. P. Andrade, R.F. Hashimoto, Growing Seed Genes from Time Series Data and Thresholded Boolean Networks with Perturbation, IEEE/ACM Trans. Comput. Biol. Bioinf., 2013. 10 (2013), 37-49. |
| [8] |
S. Kikuchi, D. Tominaga, M. Arita, K. Takahashi, M. Tomita, Dynamic modeling of genetic networks using genetic algorithm and S-system, Bioinformatics, 19 (2003), 643-650. doi: 10.1093/bioinformatics/btg027
|
| [9] |
Zhang, J., et al., Differential regulatory network-based quantification and prioritization of key genes underlying cancer drug resistance based on time-course RNA-seq data, PLOS Comput. Biol., 15 (2019), e1007435. doi: 10.1371/journal.pcbi.1007435
|
| [10] |
M. Grzegorczyk, D. Husmeier, K. D. Edwards, P. Ghazal, A. J. Millar, Modelling non-stationary gene regulatory processes with a non-homogeneous Bayesian network and the allocation sampler, Bioinformatics, 24 (2008), 2071-2078. doi: 10.1093/bioinformatics/btn367
|
| [11] |
Y. Kim, S. Han, S. Choi, D. Hwang., Inference of dynamic networks using time-course data, Briefings Bioinf., 15 (2014), 212-228. doi: 10.1093/bib/bbt028
|
| [12] |
E. Staub, J. Groene, M. Heinze, D. Mennerich, S. Roepcke, I. Klaman, et al., An expression module of WIPF1-coexpressed genes identifies patients with favorable prognosis in three tumor types, J. Mol. Med., 87 (2009), 633-644. doi: 10.1007/s00109-009-0467-y
|
| [13] |
R. N. Jorissen, P. Gibbs, M. Christie, S. Prakash, L. Lipton, J. Desai, et al., Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer, Clin. Cancer Res., 15 (2009), 7642-7651. doi: 10.1158/1078-0432.CCR-09-1431
|
| [14] |
J.. J. Smith, N. G. Deane, F. Wu, N. B. Merchant, B. Zhang, A. Jiang, et al., Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer, Gastroenterology, 138 (2010), 958-968. doi: 10.1053/j.gastro.2009.11.005
|
| [15] |
H. Chen, X. Sun, W. Ge, Y. Qian, R. Bai, S. Zheng, A seven-gene signature predicts overall survival of patients with colorectal cancer, Oncotarget, 8 (2017), 95054-95065. doi: 10.18632/oncotarget.10982
|
| [16] | Y. I. Moon, B. Rajagopalan, U. Lall, Estimation of mutual information using kernel density estimators, Phys. Rev., 52 (1995), 2318-2321. |
| [17] |
W. M. Lord, J. Sun, E. M. Bollt, Geometric k-nearest neighbor estimation of entropy and mutual information, Chaos, 28 (2018), 033114. doi: 10.1063/1.5011683
|
| [18] | T. M. Cover, J. A. Thomas, Elements of information theory, John Wiley & sons, New Jersey, 2003. |
| [19] |
H. Zhu, R. S. P. Rao, T. Zeng, L. Chen, Reconstructing dynamic gene regulatory networks from sample-based transcriptional data, Nucleic Acids Res., 40 (2012), 10657-10667. doi: 10.1093/nar/gks860
|
| [20] | The Gene Ontology Consortium., Gene Ontology Consortium: Going forward, Nucleic Acids Res., 43 (2015), D1049-D1056. |
| [21] |
D. Senft, J. Qi, Z. A. Ronai, Ubiquitin ligases in oncogenic transformation and cancer therapy, Nat. Rev. Cancer, 18 (2018), 69-88 doi: 10.1038/nrc.2017.105
|
| [22] |
A. N. Gargalionis, M. V. Karamouzis, C. Adamopoulos, A. G. Papavassiliou., Protein trafficking in colorectal carcinogenesis—targeting and bypassing resistance to currently applied treatments, Carcinogenesis, 36 (2015), 607-615. doi: 10.1093/carcin/bgv052
|
| [23] |
M. J. Pillaire, J. Selves, K. Gordien, P. A. Gouraud, C. Gentil, M. Danjoux, A 'DNA replication' signature of progression and negative outcome in colorectal cancer, Oncogene, 29 (2010), 876-887. doi: 10.1038/onc.2009.378
|
| [24] | N. Shan, W. Zhou, S. Zhang, Y. Zhang, Identification of HSPA8 as a candidate biomarker for endometrial carcinoma by using iTRAQ-based proteomic analysis, Onco. Targets Ther., 9 (2016), 2169-2179. |
| [25] | J. Samuelsson, S. Alonso, T. Ruiz-Larroya, T. H. Cheung, Y. F. Wong, M. Perucho, Frequent somatic demethylation of RAPGEF1/C3G intronic sequences in gastrointestinal and gynecological cancer, Int. J. Oncol., 38 (2011), 1575-1577. |
| [26] |
K. Honda, The biological role of actinin-4 (ACTN4) in malignant phenotypes of cancer, Cell Biosci., 5 (2015), 41. doi: 10.1186/s13578-015-0031-0
|
| [27] |
A. Calon, E. Espinet, S. Palomo-Ponce, D. V. F. Tauriello, M. Iglesias, M. V. Céspedes, et al., Dependency of Colorectal Cancer on a TGF-β-Driven Program in Stromal Cells for Metastasis Initiation, Cancer Cell, 22 (2012), 571-584. doi: 10.1016/j.ccr.2012.08.013
|
| [28] |
D. Chisanga, S. Keerthikumar, M. Pathan, D. Ariyaratne, H. Kalra, S. Boukouris, et al., Colorectal cancer atlas: An integrative resource for genomic and proteomic annotations from colorectal cancer cell lines and tissues, Nucleic Acids Res., 44 (2016), D969-D974. doi: 10.1093/nar/gkv1097
|
| [29] |
D. L. Rabosky, M. Grundler, C. Anderson, P. Title, J. J. Shi, J. W. Brown, et al., BAMM tools: An R package for the analysis of evolutionary dynamics on phylogenetic trees, Methods Ecol. Evol., 5 (2014), 701-707. doi: 10.1111/2041-210X.12199
|
| [30] |
R. Desper, J. Khan, A. A. Schäffer, Tumor classification using phylogenetic methods on expression data, J. Theor. Biol., 228 (2004), 477-496. doi: 10.1016/j.jtbi.2004.02.021
|
| [31] | Y. Wu, P. R. Bhat, T. J. Close, S. Lonardi, Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph, PLoS Genet., 4 (2008), e1000212. |
| [32] |
Y. Park, S. Shackney, R. Schwartz, Network-Based Inference of Cancer Progression from Microarray Data, IEEE/ACM Trans. Comput. Biol. Bioinf., 6 (2009), 200-212. doi: 10.1109/TCBB.2008.126
|
| [33] |
Qiu, P., A. J. Gentles, S. K. Plevritis, Discovering Biological Progression Underlying Microarray Samples, Plos Comput. Biol., 7 (2011), e1001123. doi: 10.1371/journal.pcbi.1001123
|

Figures(4) / Tables(2)

Ancheng Deng, Xiaoqiang Sun. Dynamic gene regulatory network reconstruction and analysis based on clinical transcriptomic data of colorectal cancer[J]. Mathematical Biosciences and Engineering, 2020, 17(4): 3224-3239. doi: 10.3934/mbe.2020183







 DownLoad:
DownLoad: