Citation: Vincent Carré, Sébastien Schramm, Frédéric Aubriet. Study of a complex environmental mixture by electrospray ionization and laser desorption ionization high resolution mass spectrometry: the cigarette smoke aerosol[J]. AIMS Environmental Science, 2015, 2(3): 547-564. doi: 10.3934/environsci.2015.3.547

| [1] |
Stedman RL (1968) Chemical composition of tobacco and tobacco smoke. Chem Rev 68: 153-207 doi: 10.1021/cr60252a002

|
| [2] | Smith CJ, Perfetti TA, Garg R, et al. (2003) IARC carcinogens reported in cigarette mainstream smoke and their calculated log P values. Food Chem Toxicol 41: 807-817 |
| [3] |
Dallüge J, van Stee LLP, Xu X, Williams J, et al. (2002) Unravelling the composition of very complex samples by comprehensive gas chromatography coupled to time-of-flight mass spectrometry: Cigarette smoke. J Chromatogr A 974: 169-184 doi: 10.1016/S0021-9673(02)01384-5
|
| [4] |
Takanami Y, Chida M, Hasebe H, et al. (2003) Analysis of Cigarette Smoke by an Online Thermal Desorption System and Multidimensional GC-MS. J Chromatogr Sci 41: 317-322 doi: 10.1093/chromsci/41.6.317
|
| [5] | Borgerding M, Klus H (2005) Analysis of complex mixtures -- Cigarette smoke. Exp Toxicol Pathol 57 Suppl 1: 43-73 |
| [6] |
Charles SM, Batterman SA, Jia C (2007) Composition and emissions of VOCs in main- and side-stream smoke of research cigarettes. Atmos Environ 41: 5371-5384 doi: 10.1016/j.atmosenv.2007.02.020
|
| [7] | Adam T, Mitschke S, Baker RR (2009) Investigation of tobacco pyrolysis gases and puff-by-puff resolved cigarette smoke by single photon ionisation (SPI)-time-of-flight mass spectrometry (TOFMS). Beitr Zur Tab Tob Res 23: 203-226 |
| [8] |
Wang J, Weng J-J, Jia L-Y, et al. (2012) Study on Gas Phase Components in Mainstream Cigarette Smoke by Synchrotron Radiation Photoionization Mass Spectrometry. Chin J Anal Chem 40: 1048-1052 doi: 10.1016/S1872-2040(11)60559-8
|
| [9] |
Counts ME, Hsu FS, Laffoon SW, et al. (2004) Mainstream smoke constituent yields and predicting relationships from a worldwide market sample of cigarette brands: ISO smoking conditions. Regul Toxicol Pharmacol 39: 111-134 doi: 10.1016/j.yrtph.2003.12.005
|
| [10] |
Counts ME, Morton MJ, Laffoon SW, et al. (2005) Smoke composition and predicting relationships for international commercial cigarettes smoked with three machine-smoking conditions. Regul Toxicol Pharmacol 41: 185-227 doi: 10.1016/j.yrtph.2004.12.002
|
| [11] |
Castro D, Slezakova K, Delerue-Matos C, et al. (2011) Polycyclic aromatic hydrocarbons in gas and particulate phases of indoor environments influenced by tobacco smoke: Levels, phase distributions, and health risks. Atmos Environ 45: 1799-1808 doi: 10.1016/j.atmosenv.2011.01.018
|
| [12] |
Li M, Dong J-G, Huang Z-X, et al. (2012) Analysis of Cigarette Smoke Aerosol by Single Particle Aerosol Mass Spectrometer. Chin J Anal Chem 40: 936-939 doi: 10.1016/S1872-2040(11)60555-0
|
| [13] |
Wright C (2015) Standardized methods for the regulation of cigarette-smoke constituents. Trends Anal Chem 66: 118-127 doi: 10.1016/j.trac.2014.11.011
|
| [14] |
Ding YS, Zhang L, Jain RB, et al. (2008) Levels of Tobacco-Specific Nitrosamines and Polycyclic Aromatic Hydrocarbons in Mainstream Smoke from Different Tobacco Varieties. Cancer Epidemiol Biomarkers Prev 17: 3366-3371 doi: 10.1158/1055-9965.EPI-08-0320
|
| [15] |
Ding YS, Trommel JS, Yan XJ, et al. (2005) Determination of 14 Polycyclic Aromatic Hydrocarbons in Mainstream Smoke from Domestic Cigarettes. Environ Sci Technol 39: 471-478 doi: 10.1021/es048690k
|
| [16] |
Lu X, Cai J, Kong H, et al. (2003) Analysis of Cigarette Smoke Condensates by Comprehensive Two-Dimensional Gas Chromatography/Time-of-Flight Mass Spectrometry I Acidic Fraction. Anal Chem 75: 4441-4451 doi: 10.1021/ac0264224
|
| [17] |
Lu X, Zhao M, Kong H, et al. (2004) Characterization of cigarette smoke condensates by comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry (GC×GC/TOFMS) Part 2: Basic fraction. J Sep Sci 27: 101-109 doi: 10.1002/jssc.200301659
|
| [18] |
Brokl M, Bishop L, Wright CG, et al. (2013) Analysis of mainstream tobacco smoke particulate phase using comprehensive two-dimensional gas chromatography time-of-flight mass spectrometry. J Sep Sci 36: 1037-1044 doi: 10.1002/jssc.201200812
|
| [19] |
Hughey CA, Rodgers RP, Marshall AG (2002) Resolution of 11000 Compositionally Distinct Components in a Single Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrum of Crude Oil. Anal Chem 74: 4145-4149 doi: 10.1021/ac020146b
|
| [20] | Rodgers RP, Schaub TM, Marshall AG (2005) Petroleomics: MS Returns to Its Roots. Anal Chem 77: 20-A |
| [21] |
Hertkorn N, Ruecker C, Meringer M, et al. (2007) High-precision frequency measurements: indispensable tools at the core of the molecular-level analysis of complex systems. Anal Bioanal Chem 389: 1311-1327 doi: 10.1007/s00216-007-1577-4
|
| [22] |
Kujawinski EB (2002) Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (ESI FT-ICR MS): Characterization of Complex Environmental Mixtures. Environ Forensics 3: 207-216 doi: 10.1080/713848382
|
| [23] |
Schmitt-Kopplin P, Gelencsér A, Dabek-Zlotorzynska E, et al. (2010) Analysis of the Unresolved Organic Fraction in Atmospheric Aerosols with Ultrahigh-Resolution Mass Spectrometry and Nuclear Magnetic Resonance Spectroscopy: Organosulfates As Photochemical Smog Constituents. Anal Chem 82: 8017-8026 doi: 10.1021/ac101444r
|
| [24] |
Gonsior M, Zwartjes M, Cooper WJ, et al. (2011) Molecular characterization of effluent organic matter identified by ultrahigh resolution mass spectrometry. Water Res 45: 2943-2953 doi: 10.1016/j.watres.2011.03.016
|
| [25] |
Cottrell BA, Gonsior M, Isabelle LM, et al. (2013) A regional study of the seasonal variation in the molecular composition of rainwater. Atmos Environ 77: 588-597 doi: 10.1016/j.atmosenv.2013.05.027
|
| [26] |
Cortés-Francisco N, Harir M, Lucio M, et al. (2014) High-field FT-ICR mass spectrometry and NMR spectroscopy to characterize DOM removal through a nanofiltration pilot plant. Water Res 67: 154-165 doi: 10.1016/j.watres.2014.08.046
|
| [27] |
Gonsior M, Schmitt-Kopplin P, Stavklint H, et al. (2014) Changes in Dissolved Organic Matter during the Treatment Processes of a Drinking Water Plant in Sweden and Formation of Previously Unknown Disinfection Byproducts. Environ Sci Technol 48: 12714-12722 doi: 10.1021/es504349p
|
| [28] |
Schramm S, Carré V, Scheffler J-L, et al. (2011) Analysis of Mainstream and Sidestream Cigarette Smoke Particulate Matter by Laser Desorption Mass Spectrometry. Anal Chem 83: 133-142 doi: 10.1021/ac1019842
|
| [29] |
Schramm S, Carré V, Scheffler J-L, et al. (2014) Active and passive smoking - New insights on the molecular composition of different cigarette smoke aerosols by LDI-FTICRMS. Atmos Environ 92: 411-420 doi: 10.1016/j.atmosenv.2014.04.052
|
| [30] |
Schäfer M, Drayß M, Springer A, et al. (2007) Radical Cations in Electrospray Mass Spectrometry: Formation of Open-Shell Species, Examination of the Fragmentation Behaviour in ESI-MSn and Reaction Mechanism Studies by Detection of Transient Radical Cations. Eur J Org Chem 2007: 5162-5174 doi: 10.1002/ejoc.200700199
|
| [31] |
Cole DP, Smith EA, Dalluge D, et al. (2013) Molecular characterization of nitrogen-containing species in switchgrass bio-oils at various harvest times. Fuel 111: 718-726 doi: 10.1016/j.fuel.2013.04.064
|
| [32] |
Sudasinghe N, Dungan B, Lammers P, et al. (2014) High resolution FT-ICR mass spectral analysis of bio-oil and residual water soluble organics produced by hydrothermal liquefaction of the marine microalga Nannochloropsis salina. Fuel 119: 47-56 doi: 10.1016/j.fuel.2013.11.019
|
| [33] |
Baker RR (1987) A review of pyrolysis studies to unravel reaction steps in burning tobacco. J Anal Appl Pyrolysis 11: 555-573 doi: 10.1016/0165-2370(87)85054-4
|
| [34] |
Wu Z, Rodgers RP, Marshall AG (2004) Two- and Three-Dimensional van Krevelen Diagrams: A Graphical Analysis Complementary to the Kendrick Mass Plot for Sorting Elemental Compositions of Complex Organic Mixtures Based on Ultrahigh-Resolution Broadband Fourier Transform Ion Cyclotron Resonance Mass Measurements. Anal Chem 76: 2511-2516 doi: 10.1021/ac0355449
|
| [35] |
Cho Y, Ahmed A, Islam A, et al. (2015) Developments in FT-ICR MS instrumentation, ionization techniques, and data interpretation methods for petroleomics. Mass Spectrom Rev 34: 248-263 doi: 10.1002/mas.21438
|
| [36] |
Baker RR, Bishop LJ (2004) The pyrolysis of tobacco ingredients. J Anal Appl Pyrolysis 71: 223-311 doi: 10.1016/S0165-2370(03)00090-1
|
| [37] |
Mullen CA, Boateng AA (2008) Chemical Composition of Bio-oils Produced by Fast Pyrolysis of Two Energy Crops†. Energy Fuels 22: 2104-2109 doi: 10.1021/ef700776w
|
| [38] |
Olcese R, Carré V, Aubriet F, et al. (2013) Selectivity of Bio-oils Catalytic Hydrotreatment Assessed by Petroleomic and GC*GC/MS-FID Analysis. Energy Fuels 27: 2135-2145 doi: 10.1021/ef302145g
|
| [39] |
Li S, Olegario RM, Banyasz JL, et al. (2003) Gas chromatography-mass spectrometry analysis of polycyclic aromatic hydrocarbons in single puff of cigarette smoke. J Anal Appl Pyrolysis 66: 155-163 doi: 10.1016/S0165-2370(02)00111-0
|
| [40] |
Carré V, Aubriet F, Muller J-F (2005) Analysis of cigarette smoke by laser desorption mass spectrometry. Anal Chim Acta 540: 257-268 doi: 10.1016/j.aca.2005.03.034
|
| [41] |
Aubriet F, Carré V (2010) Potential of laser mass spectrometry for the analysis of environmental dust particles—A review. Anal Chim Acta 659: 34-54 doi: 10.1016/j.aca.2009.11.047
|
| [42] |
Cho Y, Witt M, Kim YH, et al. (2012) Characterization of Crude Oils at the Molecular Level by Use of Laser Desorption Ionization Fourier-Transform Ion Cyclotron Resonance Mass Spectrometry. Anal Chem 84: 8587-8594 doi: 10.1021/ac301615m
|
| [43] |
Cho Y, Jin JM, Witt M, et al. (2013) Comparing Laser Desorption Ionization and Atmospheric Pressure Photoionization Coupled to Fourier Transform Ion Cyclotron Resonance Mass Spectrometry To Characterize Shale Oils at the Molecular Level. Energy Fuels 27: 1830-1837 doi: 10.1021/ef3015662
|
| [44] |
Le Brech Y, Delmotte L, Raya J, et al. (2015) High Resolution Solid State 2D NMR Analysis of Biomass and Biochar. Anal Chem 87: 843-847 doi: 10.1021/ac504237c
|
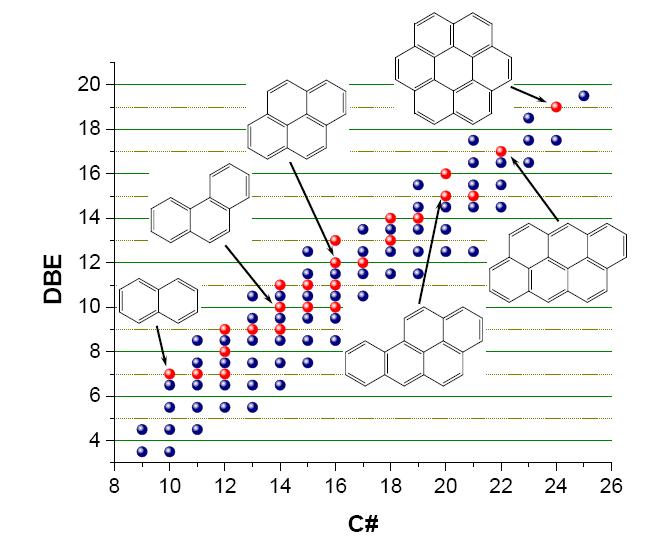
Figures(9)

Vincent Carré, Sébastien Schramm, Frédéric Aubriet. Study of a complex environmental mixture by electrospray ionization and laser desorption ionization high resolution mass spectrometry: the cigarette smoke aerosol[J]. AIMS Environmental Science, 2015, 2(3): 547-564. doi: 10.3934/environsci.2015.3.547







 DownLoad:
DownLoad: