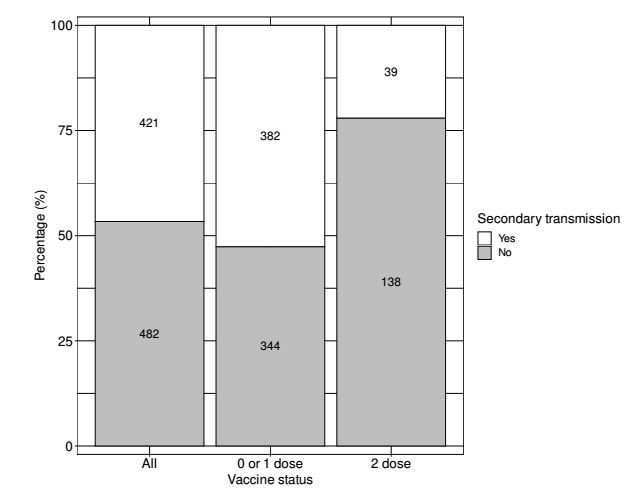
The basic reproduction number, $ R_0 $, plays a central role in measuring the transmissibility of an infectious disease, and it thus acts as the fundamental index for planning control strategies. In the present study, we apply a branching process model to meticulously observed contact tracing data from Wakayama Prefecture, Japan, obtained in early 2020 and mid-2021. This allows us to efficiently estimate $ R_0 $ and the dispersion parameter $ k $ of the wild-type COVID-19, as well as the relative transmissibility of the Delta variant and relative transmissibility among fully vaccinated individuals, from a very limited data. $ R_0 $ for the wild type of COVID-19 is estimated to be 3.78 (95% confidence interval [CI]: 3.72–3.83), with $ k = 0.236 $ (95% CI: 0.233–0.240). For the Delta variant, the relative transmissibility to the wild type is estimated to be 1.42 (95% CI: 0.94–1.90), which gives $ R_0 = 5.37 $ (95% CI: 3.55–7.21). Vaccine effectiveness, determined by the reduction in the number of secondary transmissions among fully vaccinated individuals, is estimated to be 91% (95% CI: 85%–97%). The present study highlights that basic reproduction numbers can be accurately estimated from the distribution of minor outbreak data, and these data can provide further insightful epidemiological estimates including the dispersion parameter and vaccine effectiveness regarding the prevention of transmission.
Citation: Minami Ueda, Tetsuro Kobayashi, Hiroshi Nishiura. Basic reproduction number of the COVID-19 Delta variant: Estimation from multiple transmission datasets[J]. Mathematical Biosciences and Engineering, 2022, 19(12): 13137-13151. doi: 10.3934/mbe.2022614
The basic reproduction number, $ R_0 $, plays a central role in measuring the transmissibility of an infectious disease, and it thus acts as the fundamental index for planning control strategies. In the present study, we apply a branching process model to meticulously observed contact tracing data from Wakayama Prefecture, Japan, obtained in early 2020 and mid-2021. This allows us to efficiently estimate $ R_0 $ and the dispersion parameter $ k $ of the wild-type COVID-19, as well as the relative transmissibility of the Delta variant and relative transmissibility among fully vaccinated individuals, from a very limited data. $ R_0 $ for the wild type of COVID-19 is estimated to be 3.78 (95% confidence interval [CI]: 3.72–3.83), with $ k = 0.236 $ (95% CI: 0.233–0.240). For the Delta variant, the relative transmissibility to the wild type is estimated to be 1.42 (95% CI: 0.94–1.90), which gives $ R_0 = 5.37 $ (95% CI: 3.55–7.21). Vaccine effectiveness, determined by the reduction in the number of secondary transmissions among fully vaccinated individuals, is estimated to be 91% (95% CI: 85%–97%). The present study highlights that basic reproduction numbers can be accurately estimated from the distribution of minor outbreak data, and these data can provide further insightful epidemiological estimates including the dispersion parameter and vaccine effectiveness regarding the prevention of transmission.

| [1] |
O. Diekmann, J. A. P. Heesterbeek, J. A. J. Metz, On the definition and the computation of the basic reproduction ratio $R_0$ in models for infectious diseases in heterogeneous populations, J. Math. Biol., 28 (1990), 365–382. https://doi.org/10.1007/BF00178324 doi: 10.1007/BF00178324

|
| [2] |
J. O. Lloyd-Smith, S. J. Schreiber, P. E. Kopp, W. M. Getz, Superspreading and the effect of individual variation on disease emergence, Nature, 438 (2005), 355–359. https://doi.org/10.1038/nature04153 doi: 10.1038/nature04153
|
| [3] |
J. O. Lloyd-Smith, Maximum likelihood estimation of the negative binomial dispersion parameter for highly overdispersed data, with applications to infectious diseases, PLOS ONE, 2 (2007), e180. https://doi.org/10.1371/journal.pone.0000180 doi: 10.1371/journal.pone.0000180
|
| [4] |
S. Blumberg, J. O. Lloyd-Smith, Inference of $R_0$ and transmission heterogeneity from the size distribution of stuttering chains, PLoS Comput. Biol., 9 (2013), e1002993. https://doi.org/10.1371/journal.pcbi.1002993 doi: 10.1371/journal.pcbi.1002993
|
| [5] |
H. Nishiura, P. Yan, C. K. Sleeman, C. J. Mode, Estimating the transmission potential of supercritical processes based on the final size distribution of minor outbreaks, J. Theor. Biol., 294 (2012), 48–55. https://doi.org/10.1016/j.jtbi.2011.10.039 doi: 10.1016/j.jtbi.2011.10.039
|
| [6] |
S. Zhao, M. Shen, S. S. Musa, Z. Guo, J. Ran, Z. Peng, et al., Inferencing superspreading potential using zero-truncated negative binomial model: Exemplification with COVID-19, BMC Med. Res. Methodol, 21 (2021), 30. https://doi.org/10.1186/s12874-021-01225-w doi: 10.1186/s12874-021-01225-w
|
| [7] |
D. C. Adam, P. Wu, J. Y. Wong, E. H. Y. Lau, T. K. Tsang, S. Cauchemez, et al., Clustering and superspreading potential of SARS-CoV-2 infections in Hong Kong, Nat. Med., 26 (2020), 1714–1719. https://doi.org/10.1038/s41591-020-1092-0 doi: 10.1038/s41591-020-1092-0
|
| [8] |
A. Endo, Centre for the Mathematical Modelling of Infectious Diseases COVID-19 Working Group, S. Abbott, A. J. Kucharski, S. Funk, Estimating the overdispersion in COVID-19 transmission using outbreak sizes outside China, Wellcome Open Res., 5 (2020), 67. https://doi.org/10.12688/wellcomeopenres.15842.3 doi: 10.12688/wellcomeopenres.15842.3
|
| [9] |
H. Lee, C. Han, J. Jung, S. Lee, Analysis of superspreading potential from transmission clusters of COVID-19 in South Korea, Int. J. Environ. Res. Public Health, 18 (2021), 12893. https://doi.org/10.3390/ijerph182412893 doi: 10.3390/ijerph182412893
|
| [10] |
Y. Zhang, Y. Li, L. Wang, M. Li, X. Zhou, Evaluating transmission heterogeneity and super-spreading event of COVID-19 in a metropolis of China, Int. J. Environ. Res. Public Health, 17 (2020), 3705. https://doi.org/10.3390/ijerph17103705 doi: 10.3390/ijerph17103705
|
| [11] |
A. Tariq, Y. Lee, K. Roosa, S. Blumberg, P. Yan, S. Ma, et al., Real-time monitoring the transmission potential of COVID-19 in Singapore, March 2020, BMC Medicine, 18 (2020), 166. https://doi.org/10.1186/s12916-020-01615-9 doi: 10.1186/s12916-020-01615-9
|
| [12] |
L. Wang, X. Didelot, J. Yang, G. Wong, Y. Shi, W. Liu, et al., Inference of person-to-person transmission of COVID-19 reveals hidden super-spreading events during the early outbreak phase, Nat. Commun., 11 (2020), 5006. https://doi.org/10.1038/s41467-020-18836-4 doi: 10.1038/s41467-020-18836-4
|
| [13] |
K. Nakajo, H. Nishiura, Transmissibility of asymptomatic COVID-19: Data from Japanese clusters, Int. J. Infect. Dis., 105 (2021), 236–238. https://doi.org/10.1016/j.ijid.2021.02.065 doi: 10.1016/j.ijid.2021.02.065
|
| [14] |
H. Hwang, J. S. Lim, S. A. Song, C. Achangwa, W. Sim, G. Kim, et al., Transmission dynamics of the Delta variant of SARS-CoV-2 infections in South Korea, J. Infect. Dis., 225 (2022), 793–799. https://doi.org/10.1093/infdis/jiab586 doi: 10.1093/infdis/jiab586
|
| [15] |
J. Middleton, H. Lopes, K. Michelson, J. Reid, Planning for a second wave pandemic of COVID-19 and planning for winter: A statement from the Association of Schools of Public Health in the European Region, Int. J. Public Health, 65 (2020), 1525–1527. https://doi.org/10.1007/s00038-020-01455-7 doi: 10.1007/s00038-020-01455-7
|
| [16] |
S. X. Zhang, F. Arroyo Marioli, R. Gao, S. Wang, A second wave? What do people mean by COVID waves? – a working definition of epidemic waves, RMHP, 14 (2021), 3775–3782. https://doi.org/10.2147/RMHP.S326051 doi: 10.2147/RMHP.S326051
|
| [17] |
R. Kinoshita, S. M. Jung, T. Kobayashi, A. R. Akhmetzhanov, H. Nishiura, Epidemiology of coronavirus disease 2019 (COVID-19) in Japan during the first and second waves, Math. Biosci. Eng., 19 (2022), 6088–6101. https://doi.org/10.3934/mbe.2022284 doi: 10.3934/mbe.2022284
|
| [18] |
T. Kuniya, Evaluation of the effect of the state of emergency for the first wave of COVID-19 in Japan, Infect. Dis. Model., 5 (2020), 580–587. https://doi.org/10.1016/j.idm.2020.08.004 doi: 10.1016/j.idm.2020.08.004
|
| [19] | Digital Agency, Japan, [COVID-19 vaccination status], 2022. Avaliable from: https://info.vrs.digital.go.jp/dashboard/ |
| [20] |
P. Whittle, The outcome of a stochastic epidemic —a note on Bailey's paper, Biometrika, 42 (1955), 116–122. https://doi.org/10.1093/biomet/42.1-2.116 doi: 10.1093/biomet/42.1-2.116
|
| [21] |
W. Tritch, L. J. Allen, Duration of a minor epidemic, Infect. Dis. Model., 3 (2018), 60–73. https://doi.org/10.1016/j.idm.2018.03.002 doi: 10.1016/j.idm.2018.03.002
|
| [22] | P. Yan, G. Chowell, Quantitative Methods for Investigating Infectious Disease Outbreaks, Springer, 2019. |
| [23] |
K. Sneppen, B. F. Nielsen, R. J. Taylor, L. Simonsen, Overdispersion in COVID-19 increases the effectiveness of limiting nonrepetitive contacts for transmission control, Proc. Natl. Acad. Sci., 118 (2021), e2016623118. https://doi.org/10.1073/pnas.2016623118 doi: 10.1073/pnas.2016623118
|
| [24] |
Q. Bi, Y. Wu, S. Mei, C. Ye, X. Zou, Z. Zhang, et al., Epidemiology and transmission of COVID-19 in 391 cases and 1286 of their close contacts in Shenzhen, China: A retrospective cohort study, Lancet Infect. Dis., 20 (2020), 911–919. https://doi.org/10.1016/S1473-3099(20)30287-5 doi: 10.1016/S1473-3099(20)30287-5
|
| [25] |
R. Laxminarayan, B. Wahl, S. R. Dudala, K. Gopal, C. Mohan B, S. Neelima, et al., Epidemiology and transmission dynamics of COVID-19 in two Indian states, Science, 370 (2020), 691–697. https://doi.org/10.1126/science.abd7672 doi: 10.1126/science.abd7672
|
| [26] |
K. Sun, W. Wang, L. Gao, Y. Wang, K. Luo, L. Ren, et al., Transmission heterogeneities, kinetics, and controllability of SARS-CoV-2, Science, 371 (2021), eabe2424. https://doi.org/10.1126/science.abe2424 doi: 10.1126/science.abe2424
|
| [27] |
J. Riou, C. L. Althaus, Pattern of early human-to-human transmission of Wuhan 2019 novel coronavirus (2019-nCoV), December 2019 to January 2020, Eurosurveillance, 25 (2020), 2000058, https://doi.org/10.2807/1560-7917.ES.2020.25.4.2000058 doi: 10.2807/1560-7917.ES.2020.25.4.2000058
|
| [28] | R Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2020. Available from: https://www.R-project.org/. |
| [29] |
M. A. Billah, M. M. Miah, M. N. Khan, Reproductive number of coronavirus: A systematic review and meta-analysis based on global level evidence, PLOS ONE, 15 (2020), e0242128. https://doi.org/10.1371/journal.pone.0242128 doi: 10.1371/journal.pone.0242128
|
| [30] |
Y. Liu, A. A. Gayle, A. Wilder-Smith and J. Rocklöv, The reproductive number of COVID-19 is higher compared to SARS coronavirus, Journal of Travel Medicine, 27 (2020), taaa021. https://doi.org/10.1093/jtm/taaa021. doi: 10.1093/jtm/taaa021
|
| [31] |
Y. Liu, J. Rocklöv, The reproductive number of the Delta variant of SARS-CoV-2 is far higher compared to the ancestral SARS-CoV-2 virus, J. Travel Med., 28 (2021), taab124. https://doi.org/10.1093/jtm/taab124 doi: 10.1093/jtm/taab124
|
| [32] |
M. Zhang, J. Xiao, A. Deng, Y. Zhang, Y. Zhuang, T. Hu, et al., Transmission dynamics of an outbreak of the COVID-19 Delta variant B.1.617.2 — Guangdong Province, China, May–June 2021, CCDCW, 3 (2021), 584–586. https://doi.org/10.46234/ccdcw2021.148 doi: 10.46234/ccdcw2021.148
|



Figures(4)

Minami Ueda, Tetsuro Kobayashi, Hiroshi Nishiura. Basic reproduction number of the COVID-19 Delta variant: Estimation from multiple transmission datasets[J]. Mathematical Biosciences and Engineering, 2022, 19(12): 13137-13151. doi: 10.3934/mbe.2022614







 DownLoad:
DownLoad: