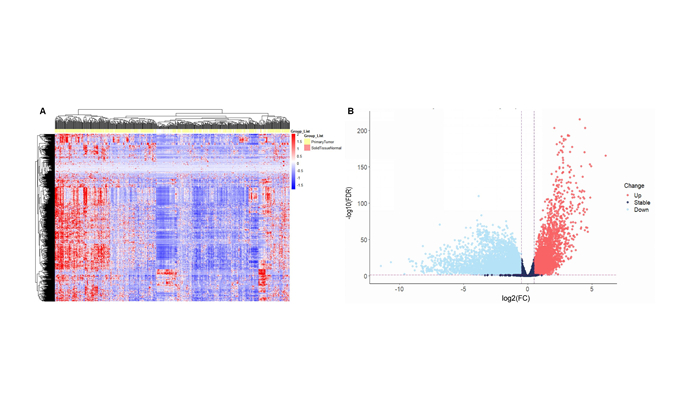
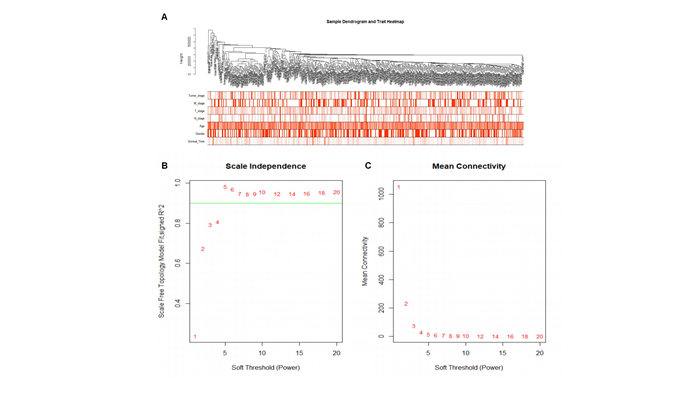
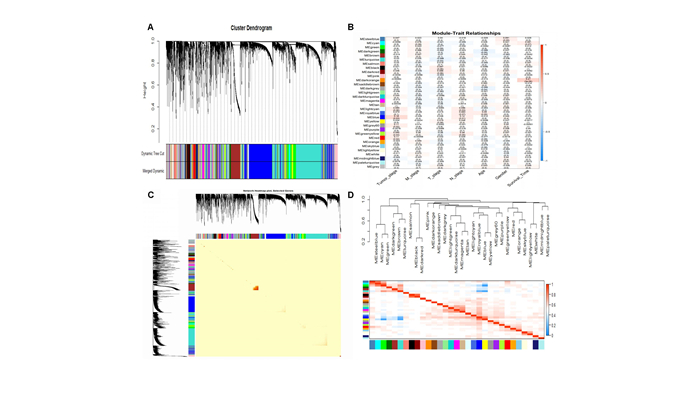
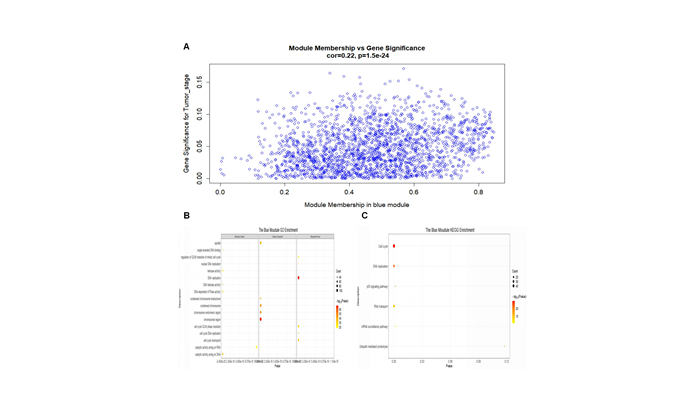
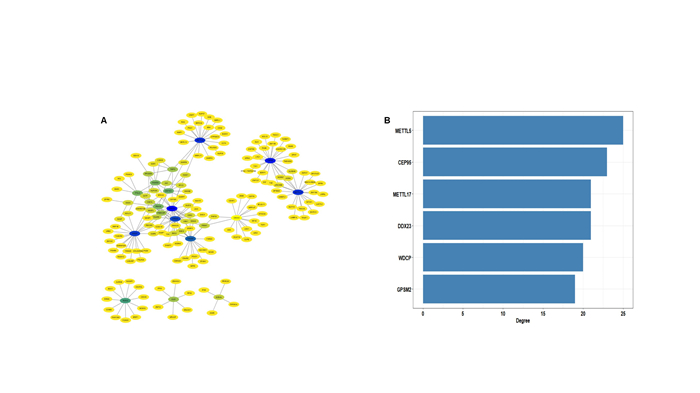
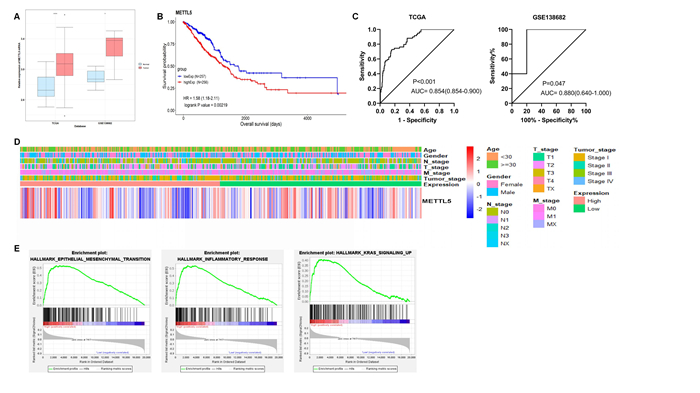
Lung adenocarcinoma (LUAD) is a frequently diagnosed malignant tumor that is highly invasive and lethal. The prognosis of patients with LUAD still needs to be improved, as conventional treatment is remarkably well tolerated. In this study, the expression profile of LUAD in the TCGA database was used for differential expression analysis, and differential expression genes were determined to construct a weighted gene co-expression network analysis (WGCNA) for dividing and finding the gene modules with the highest correlation with tumor stage. Here, METTL5, DDX23, GPSM2, CEP95, WDCP, and METL17 were identified as hub genes. According to the relation degree, METTL5 was determined as the candidate gene in this study. Difference analysis and receiver operating characteristic (ROC) curve were applied to identify the predictive performance of METTL5 in LUAD, and Kaplan-Meier (KM) analysis showed that the prognosis of LUAD patients with high METTL5 expression was poor. Further GSEA analysis showed that high-expressed METTL5 was related to epithelial-mesenchymal transition and other pathways. Therefore, METTL5 may be involved in the occurrence and malignant progression of LUAD. The current findings provide an effective molecular target for early diagnosis of LUAD, helping monitor the malignant progression of LUAD and improve the prognosis of LUAD patients.
Citation: Xinwang Yan, Xiaowen Zhao, Qing Yan, Ye Wang, Chunling Zhang. Analysis of the role of METTL5 as a hub gene in lung adenocarcinoma based on a weighted gene co-expression network[J]. Mathematical Biosciences and Engineering, 2021, 18(5): 6608-6619. doi: 10.3934/mbe.2021327
Lung adenocarcinoma (LUAD) is a frequently diagnosed malignant tumor that is highly invasive and lethal. The prognosis of patients with LUAD still needs to be improved, as conventional treatment is remarkably well tolerated. In this study, the expression profile of LUAD in the TCGA database was used for differential expression analysis, and differential expression genes were determined to construct a weighted gene co-expression network analysis (WGCNA) for dividing and finding the gene modules with the highest correlation with tumor stage. Here, METTL5, DDX23, GPSM2, CEP95, WDCP, and METL17 were identified as hub genes. According to the relation degree, METTL5 was determined as the candidate gene in this study. Difference analysis and receiver operating characteristic (ROC) curve were applied to identify the predictive performance of METTL5 in LUAD, and Kaplan-Meier (KM) analysis showed that the prognosis of LUAD patients with high METTL5 expression was poor. Further GSEA analysis showed that high-expressed METTL5 was related to epithelial-mesenchymal transition and other pathways. Therefore, METTL5 may be involved in the occurrence and malignant progression of LUAD. The current findings provide an effective molecular target for early diagnosis of LUAD, helping monitor the malignant progression of LUAD and improve the prognosis of LUAD patients.

| [1] |
F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre, A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J Clin., 68 (2018), 394-424. doi: 10.3322/caac.21492. doi: 10.3322/caac.21492

|
| [2] | J. A. Barta, C. A. Powell, J. P. Wisnivesky, Global Epidemiology of Lung Cancer, Ann. Glob. Health, 85 (2019), 8. doi: 10.5334/aogh.2419 |
| [3] |
M. C. S. Wong, X. Q. Lao, K. F. Ho, W. B. Goggins, S. L. A. Tse, Incidence and mortality of lung cancer:global trends and association with socioeconomic status, Sci. Rep., 7 (2017), 1-9. doi: 10.1038/s41598-016-0028-x
|
| [4] | X. Hua, W. Zhao, A. C. Pesatori, D. Consonni, N. E. Caporaso, T. Zhang, et al., Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma, Nat. Commun., 11 (2020), 1-11. |
| [5] |
T. V. Denisenko, I. N. Budkevich, B. Zhivotovsky, Cell death-based treatment of lung adenocarcinoma, Cell Death Dis., 9 (2018), 1-14. doi: 10.1038/s41419-017-0012-9
|
| [6] | R. Liu, W. Zhang, Z. Q. Liu, H. H. Zhou, Associating transcriptional modules with colon cancer survival through weighted gene co-expression network analysis, BMC Genom., 18 (2017), 361. |
| [7] | B. Zhang, S. Horvath, A general framework for weighted gene co-expression network analysis, Stat. Appl. Genet. Mol. Biol., 4 (2005), 17. doi: 10.2202/1544-6115.1128. |
| [8] | K. Tomczak, P. Czerwińska, M. Wiznerowicz, The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge, Contemp. Oncol., 19 (2015), A68. |
| [9] |
J. Liu, T. Lichtenberg, K. A. Hoadley, L. M. Poisson, A. J. Lazar, A. D. Cherniack, et al., An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics, Cell, 173 (2018), 400-416. e11. doi: 10.1016/j.cell.2018.02.052
|
| [10] | G. P. Kalemkerian, N. Narula, E. B. Kennedy, W. A. Biermann, J. Donington, N. B. Leighl, et al., Molecular testing guideline for the selection of patients with lung cancer for treatment with targeted tyrosine kinase inhibitors: American society of clinical oncology endorsement of the college of american pathologists/international association for the study of lung cancer/association for molecular pathology clinical practice guideline update, J. Clin. Oncol., 36 (2018), 911. |
| [11] | D. J. Myers, J. M Wallen, Lung adenocarcinoma, StatPearls[Internet], 2020. |
| [12] |
J. Zhang, Q. Dai, D. Park, X. Deng, Targeting DNA replication stress for cancer therapy, Genes, 7 (2016), 51. doi: 10.3390/genes7080051
|
| [13] |
H. Kitao, M. Iimori, Y. Kataoka, T. Wakasa, E. Tokunaga, H. Saeki, et al., DNA replication stress and cancer chemotherapy, Cancer Sci., 109 (2018), 264-271. doi: 10.1111/cas.13455
|
| [14] | J. V. Forment, M. J. O'Connor, Targeting the replication stress response in cancer, Pharmacol. Ther., 188 (2018), 155-167. |
| [15] | S. Sun, K. Fei, G. Zhang, J. Wang, Y. Yang, W. Guo, et al., Construction and comprehensive analyses of a METTL5-associated prognostic signature with immune implication in lung adenocarcinomas, Front. Genet., 11 (2021), 1801. |
| [16] |
M. K. Jolly, C. Ward, M. S. Eapen, S. Myers, O. Hallgren, H. Levine, et al., Epithelial--mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease, Dev. Dyn., 247 (2018), 346-358. doi: 10.1002/dvdy.24541
|
| [17] |
K. Li, D. Sun, Q. Gou, X. Ke, Y. Gong, Y. Zuo, et al., Long non-coding RNA linc00460 promotes epithelial-mesenchymal transition and cell migration in lung cancer cells, Cancer Lett., 420 (2018), 80-90. doi: 10.1016/j.canlet.2018.01.060
|
| [18] | Y. Lou, L. Diao, E. R. P. Cuentas, W. L. Denning, L. Chen, Y. H. Fan, et al., Epithelial--mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma, Clin. Cancer Res., 22 (2018), 3630-3642. |
| [19] |
H. Ji, A. M. Houghton, T. J. Mariani, S. Perera, C. B. Kim, R. Padera, et al., K-ras activation generates an inflammatory response in lung tumors, Oncogene, 25 (2006), 2105-2112. doi: 10.1038/sj.onc.1209237
|
| [20] | M. Serresi, B. Siteur, D. Hulsman, C. Company, M. J. Schmitt, C. Lieftink, et al., Ezh2 inhibition in Kras-driven lung cancer amplifies inflammation and associated vulnerabilities, J. Exp. Med., 215 (2018), 3115-3135. |
| [21] | J. F. Costello, C. Plass, Methylation matters, J. Med. Genet., 38 (2001), 285-303. |
| [22] |
T. Sun, R. Wu, L. Ming, The role of m6A RNA methylation in cancer, Biomed. Pharmacother., 112 (2019), 108613. doi: 10.1016/j.biopha.2019.108613
|
| [23] | Y. Pan, P. Ma, Y. Liu, W. Li, Y. Shu, Multiple functions of m6A RNA methylation in cancer, J. Hematol. Oncol., 11 (2018), 48. |
| [24] |
Q. Cui, H. Shi, P. Ye, L. Li, Q. Qu, G. Sun, et al., m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells, Cell Rep., 18 (2017), 2622-2634. doi: 10.1016/j.celrep.2017.02.059
|
| [25] |
V. V. Ignatova, P. Stolz, S. Kaiser, T. H. Gustafsson, P. R. Lastres, A. Sanz-Moreno, et al., The rRNA m6A methyltransferase METTL5 is involved in pluripotency and developmental programs, Genes Dev., 34 (2020), 715-729. doi:10.1101/gad.333369.119. doi: 10.1101/gad.333369.119
|
| [26] | H. Chen, Q. Liu, D. Yu, S. K. Natchiar, C. Zhou, C. H. Hsu, et al., METTL5, an 18S rRNA-specific m6A methyltransferase, modulates expression of stress response genes, Bio. Rxiv., doi: (2020)10.1101/2020.0427.064162. |
| [27] |
V. V. Ignatova, P. Stolz, S. Kaiser, T. H. Gustafsson, P. R. Lastres, A. Sanz-Moreno, et al., The rRNA m6A methyltransferase METTL5 is involved in pluripotency and developmental programs, Genes Dev., 34 (2020), 715-729. doi: 10.1101/gad.333369.119
|
Figures(6)

Xinwang Yan, Xiaowen Zhao, Qing Yan, Ye Wang, Chunling Zhang. Analysis of the role of METTL5 as a hub gene in lung adenocarcinoma based on a weighted gene co-expression network[J]. Mathematical Biosciences and Engineering, 2021, 18(5): 6608-6619. doi: 10.3934/mbe.2021327







 DownLoad:
DownLoad: