Citation: Pedro José Gutiérrez-Diez, Jose Russo. Design of personalized cancer treatments by use of optimal control problems: The case of chronic myeloid leukemia[J]. Mathematical Biosciences and Engineering, 2020, 17(5): 4773-4800. doi: 10.3934/mbe.2020261

| [1] | A. J. Lotka, Contribution to the Theory of Periodic Reaction, J. Phys. Chem., 14 (1910), 271-274. |
| [2] | A. J. Lotka, Analytical Note on Certain Rhythmic Relations in Organic Systems, Proc. Natl. Acad. Sci. U.S.A., 6 (1920), 410-415. |
| [3] | A. J. Lotka, Elements of Physical Biology, Williams and Wilkins, 1925. |
| [4] | V. Volterra, Variazioni e fluttuazioni del numero d'individui in specie animali conviventi, Mem. R. Accad. Naz. Lincei, 2 (1926), 31-113. |
| [5] | V. Volterra, Fluctuations in the abundance of a species considered mathematically, Nature, 118, (1926), 558-560. |
| [6] | V. Volterra, Leçons sur la théorie mathématique de la lutte pour la vie, Paris: Gauthier-Villars, 1931. |
| [7] | L. A. Loeb, K. R. Loeb, J. P. Anderson, Multiple mutations and cancer, Proc. Natl. Acad. Sci. U.S.A., 100 (2003), 776-781. |
| [8] | G. Magombedze, W. Garira, E. Mwenje, C. P. Bhunu, Optimal control for HIV-1 multi-drug therapy, Int. J. Comp. Math., 88 (2011), 314-340. |
| [9] | N. Tarfulea, A mathematical model for HIV treatment with time-varying antiretroviral therapy, Int. J. Comp. Math., 88, (2011), 3217-3235. |
| [10] | Y. Koizumi, S. Iwami, Mathematical modeling of multi-drugs therapy: A challenge for determining the optimal combinations of antiviral drugs, Theor. Biol. Med. Mod., 11 (2014), 41. |
| [11] | H. T. Banks, Modeling and Control in the Biomedical Sciences, Springer, 1975. |
| [12] | J. D. Murray, Mathematical Biology I: An Introduction, Springer, 2002. |
| [13] | J. D. Murray, Mathematical Biology II: Spatial Models and Biomedical Applications, Springer, 2003. |
| [14] | C. W. Clark, Mathematical Bioeconomics. The Optimal Management of Renewable Resources, John Wiley & Sons, Inc., 1990. |
| [15] | U. Ledzewicz, H. Schättler, A. Friedman, E. Kashdan, Mathematical methods and models in Biomedicine, Springer, 2013. |
| [16] | M. Eisen, Mathematical Methods and Models in the Biological Sciences, Prentice Hall, 1988. |
| [17] | P. J. Gutiérrez Diez, I. H. Russo, J. Russo, The Evolution of the Use of Mathematics in Cancer Research, Springer, 2012. |
| [18] | A. Świerniak, U. Ledzewicz, H. Schättler, Optimal Control for a Class of Compartmental Models in Cancer Chemotherapy, Int. J. Appl. Math. Comp. Sci., 13 (2003), 357-368. |
| [19] | J. M. Murray, Optimal control for a cancer chemotherapy problem with general growth and cost functions, Math. Biosci. 98 (1990), 273-287. |
| [20] | J. M. Murray, Some optimal control problems in cancer chemotherapy with a toxicity limit, Math. Biosci., 100 (1990), 49-67. |
| [21] | S. Nanda, H. Moore, S. Lenhart, Optimal control of treatment in a mathematical model of chronic myelogenous leukemia, Math. Biosci., 210 (2007), 143-156. |
| [22] | B. E. Aïnseba, C. Benosman, Optimal control for resistance and suboptimal response in CML, Math. Biosci., 227 (2010), 81-93. |
| [23] |
K. Schepers, E. M. Pietras, D. Reynaud, J. Flach, M. Binnewies, T. Garg, et al., Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche, Cell Stem Cell, 13 (2013), 285-299. doi: 10.1016/j.stem.2013.06.009

|
| [24] | S. Kapoor, V. P. Rallabandi, C. Sakode, R. Padhi, P.K. Roy, A patient-specific therapeutic approach for tumour cell population extinction and drug toxicity reduction using control systems-based dose-profile design, Theor. Biol. Med. Mod., 10 (2013), 68. |
| [25] | P. J. Gutiérrez-Diez, M. Á.,López-Marcos, J. Martínez-Rodríuez, J. Russo, The effects of time valuation in cancer optimal therapies: A study of chronic myeloid leukemia, Theor. Biol. Med. Mod., 16 (2019), 10. |
| [26] | M. Baccarani, F. Castagnetti, G. Gugliotta, G. Rosti, A review of the European LeukemiaNet recommendations for the management of CML, Ann. Hematol., 94 (2015), S141-S147. |
| [27] | M. W. Deininger, J. G. Hodgson, N. P. Shah, J. E. Cortes, D. W. Kim, F. E. Nicolini, et al., Compound mutations in BCR-ABL1 are not major drivers of primary or secondary resistance to ponatinib in CP-CML patients, Blood, 127 (2016), 703-712. |
| [28] | F. Michor, T. P. Hughes, Y. Iwasa, S. Branford, N. P. Shah, C. L. Sawyers, et al., Dynamics of chronic myeloid leukaemia, Nature, 435 (2005), 1267-1270. |
| [29] | D. Murray Lyon, Does the reaction to adrenalin obey Weber's Law?, J. Pharmacol. Exp. Ther., 21 (1923), 229-235. |
| [30] | M. Inoue, K. Kaneko, Weber's law for biological responses in autocatalytic networks of chemical reactions, Phys. Rev. Lett., 107 (2011), 048301. |
| [31] | H. W. Sinn, Weber's law and the biological evolution of risk preferences: The selective dominance of the logarithmic utility function, Gen. Pap. Risk Ins. Theor., 28 (2002), 87-100. |
| [32] | R. Capdeville, S. Silverman, Imatinib: A targeted clinical drug development, Semin. Hematol., 40 (2003), 15-20. |
| [33] | M. Bonifacio, F. Stagno, L. Scaffidi, M. Krampera, F. Di Raimondo, Management of Chronic Myeloid Leukemia in Advanced Phase, Front. Oncol., 9 (2019), 1132. |
| [34] | S. Soverini, A. Hochhaus, F. E. Nicolini, F. Gruber, T. Lange, G. Saglio, et al., BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: Recommendations from an expert panel on behalf of European LeukemiaNet. Blood, 118 (2011), 1208-1215. |
| [35] | I. Galinsky, S. Buchanan, Guide to Interpreting Disease Responses in Chronic Myeloid Leukemia, J. Adv. Prac. Oncol., 3, (2012), 225-236. |
| [36] | S. Prabhu, D. Saadat, M. Zhang, L. Halbur, J. P. Fruehauf, S. T. Ong, A novel mechanism for BCR-ABL action: BCR-ABL-mediated induction of the eIF4F translation initiation complex and mRNA translation, Oncogene, 26 (2007), 1188-1200. |
| [37] | L. Hu, L. Pu, D. Yang, C. Zhang, H. Wang, Y. Ding, et al., How to detect the rare BCR-ABL (e14a3) transcript: A case report and literature review, Oncol. Lett., 14 (2017), 5619-5623. |
| [38] | S. I. Ismail, R. G. Naffa, A. F. Yousef, M. T. Ghanim, Incidence of BCR-ABL fusion transcripts in healthy individuals, Mol. Med. Rep. 9 (2014), 1271-1276. |
| [39] |
T. Hughes, G. Saglio, A. Quintás-Cardama, M. J. Mauro, D. W. Kim, J. H. Lipton, et al., BCR-ABL1 mutation development during first-line treatment with dasatinib or imatinib for chronic myeloid leukemia in chronic phase, Leukemia, 29 (2015), 1832-1838. doi: 10.1038/leu.2015.168
|
| [40] | J. Kaeda, D. O'Shea, R. M. Szydlo, E. Olavarria, F. Dazzi, D. Marin, et al., Serial measurement of BCR-ABL transcripts in the peripheral blood after allogeneic stem cell transplantation for chronic myeloid leukemia: An attempt to define patients who may not require further therapy, Blood, 107 (2006), 4171-4176. |
| [41] | M. Houshmand, G. Simonetti, P. Circosta, V. Gaidano, A. Cignetti, G. Martinelli, et al., Chronic myeloid leukemia stem cells, Leukemia, 33 (2019), 1543-1556. |
| [42] | F. Loscocco, G. Visani, S. Galimberti, A. Curti, A. Isidori, BCR-ABL Independent Mechanisms of Resistance in Chronic Myeloid Leukemia, Front. Oncol., 9 (2019), 939. |
| [43] | H. Kitamura, Y. Tabe, T. Ai, K. Tsuchiya, M. Yuri, S. Misawa, et al., A new highly sensitive real-time quantitative-PCR method for detection of BCR-ABL1 to monitor minimal residual disease in chronic myeloid leukemia after discontinuation of imatinib, PLoS One, 14 (2019), e0207170. |
| [44] | R. Arora, R. D. Press, Measurement of BCR-ABL1 transcripts on the International Scale in the United States: Current status and best practices, Leuk. Lymphoma, 58 (2017), 8-16. |
| [45] | Physycians' Desk Reference, PDR Network (ed) 64 edition, LLC at Montvale, 2010. |
| [46] | A. Quintás-Cardama, J. Cortes, H. Kantarjian, Biology of Chronic and Acute Myeloid Leukemia, in The Molecular Basis of Cancer, Elsevier, (2008), 371-383. |
| [47] | L. Löf, L. Arngården, U. Olsson-Strömberg, B. Siart, M. Jansson, J. S. Dahlin, et al., Flow Cytometric Measurement of Blood Cells with BCR-ABL1 Fusion Protein in Chronic Myeloid Leukemia, Sci. Rep. 7 (2017), 623. |
| [48] | D. Raspadori, P. Pacelli, A. Sicuranza, E. Abruzzese, A. Iurlo, D. Cattaneo, et al., Flow Cytometry Assessment of CD26+ Leukemic Stem Cells in Peripheral Blood: A Simple and Rapid New Diagnostic Tool for Chronic Myeloid Leukemia, Cytometry Part B, 96 (2019), 294-299. |
| [49] | F. E. Craig, K. A. Foon, Flow cytometric immunophenotyping for hematologic neoplasms, Blood, 111 (2008), 3941-3967. |
| [50] | D. Campana, Minimal residual disease in acute lymphoblastic leukemia, Semin. Hematol., 46 (2009), 100-106. |
| [51] | D. Campana, Role of minimal residual disease monitoring in adult and pediatric acute lymphoblastic leukemia, Haematol./Oncol. Clin. N.A., 23 (2009), 1083-1098. |
| [52] | N. Shah,Medical management of CML, Hematology, 1 (2007), 371-375. |
| [53] | P. K. Bhamidipati, H. Kantarjian, J. Cortes, A. M. Cornelison, E. Jabbour, Management of imatinib-resistant patients with chronic myeloid leukemia, Ther. Adv. Hematol., 4 (2013), 103-117. |
| [54] | M. S. Marcolino, E. Boersma, N. C. D. Clementino, A. V. Macedo, A. D. Marx-Neto, M. H. C. R. Silva, et al., Imatinib treatment duration is related to decreased estimated glomerular filtration rate in chronic myeloid leukemia patients, Ann. Oncol., 22 (2011), 2073-2079. |
| [55] | M. Baccarani, J. Cortes, F. Pane, D. Niederwieser, G. Saglio, J. Apperley, et al., Chronic myeloid leukemia: An update of concepts and management recommendations of European LeukemiaNet, J. Clin. Oncol., 27 (2009), 6041-6051. |
| [56] | R. A. Everett, Y. Zhao, K. B. Flores, Y. Kuang, Data and implication based comparison of two chronic myeloid leukemia models, Math. Biosci. Eng., 10 (2013), 1501-1518. |
| [57] | C. Fava, G. Saglio, Can we and should we improve on frontline imatinib therapy for chronic myeloid leukemia?, Semin. Hematol., 47 (2010), 319-326. |
| [58] | P. Valent, Imatinib-resistant chronic myeloid leukemia (CML): Current concepts on pathogenesis and new emerging pharmacologic approaches, Biol. Targets Ther., 1 (2007), 433-448. |
| [59] | R. Bhatia, Chronic Myeloid Leukemia, in Hematology, seventh edition, Elsevier, (2018), 1055-1070. |
| [60] | T. R. Randolph, Myeloproliferative neoplasms, in Rodak's Hematology Clinical Applications and Principles, St. Louis, Missouri: Saunders, (2015), 561-590. |
| [61] | N. Bouizem, M. Helal, B. Aïnseba, A. Lakmeche, Leukemia mathematical model, ITM Web Conf., 4 (2015), 01006. |
| [62] | A. Hochhaus, S. Kreil, A. S. Corbin, P. La Rosée, M. C. Müller, T. Lahaye, et al., Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy, Leukemia, 16 (2002), 2190-2196. |
| [63] | V. Karavasilis, A. Reid, R. Sinha, J. S. De Bono, Cancer drug resistance, in Cancer Drug Design and Discovery, (S. Neidle ed.), (2008), Elsevier. |
| [64] | M. Baccarani, G. Saglio, J. Goldman, A. Hochhaus, B. Simonsson, F. Appelbaum, et al., Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European Leukemia Net, Blood, 108 (2006), 1809-1820. |
| [65] | R.P. Nelson Jr, K. Cornetta, K. E. Ward, S. Ramanuja, C. Fausel, L.D. Cripe, Desensitization to imatinib in patients with leukemia, Ann. Allergy, Asthma, Immunol., 97 (2006), 216-222. |
| [66] | M. Albayrak, H. Celebi, A. Albayrak, E. S. Can, V. Aslan, B. Onec, et al., Serious skin reaction associated with imatinib in a patient with chronic myeloid leukemia, Eurasian J. Med., 43 (2011), 192-195. |
| [67] | V. Chou, S. McClelland, D. Resnick, M. Lee-Wong, Successful Desensitization of an Adult with Type I Hypersensitivity to Imatinib, Internet J. Asthma, Allergy Immunol., 4 (2004), 2. |
| [68] | E. Faber, M. Divoká, I. Skoumalová, M. Novák, I. Marešová, K. Mičová, et al., A lower dosage of imatinib is sufficient to maintain undetectable disease in patients with chronic myeloid leukemia with long-term low-grade toxicity of the treatment, Leuk. Lymphoma, 57 (2016), 370-375. |
| [69] | Y. Zhu, S. X. Qian, Clinical efficacy and safety of imatinib in the management of Ph(+) chronic myeloid or acute lymphoblastic leukemia in Chinese patients, OncoTargets Ther., 7 (2014), 395-404. |
| [70] | European Leukemia Net, 2020. Available from: https://www.leukemia-net.org/content/leukemias/cml/euro_and_sokal_score/index_eng.html. |
| [71] | L. C. Kuntegowdanahalli, G. B. Kanakasetty, A. H. Thanky, L. Dasappa, L. A. Jacob, S. B. Mallekavu, et al., Prognostic and predictive implications of Sokal, Euro and EUTOS scores in chronic myeloid leukaemia in the imatinib era-experience from a tertiary oncology centre in Southern India, Ecancermedicalscience, 10 (2016), 679. |
| [72] | S. Chhikara, S. Sazawal, K. Singh, R. Chaubey, H. Pati, S. Tyagi, et al., Comparative analysis of the Sokal, Euro and European Treatment and Outcome Study score in prognostication of Indian chronic myeloid leukemia-chronic phase patients on imatinib, S.A. J. Cancer, 7, (2018), 258-262. |
| [73] |
J. Hasford, M. Baccarani, V. Hoffmann, J. Guilhot, S. Saussele, G. Rosti, et al., Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score, Blood, 118 (2011), 686-692. doi: 10.1182/blood-2010-12-319038
|
| [74] | M. Baccarani, F. Castagnetti, G. Gugliotta, G. Rosti, A review of the European Leukemia Net recommendations for the management of CML, Ann. Hematol., 94 (2015), S141-S147. |
| [75] | M. Schemionek, T. Spieker, L. Kerstiens, C. Elling, M. Essers, A. Trumpp, et al., Leukemic spleen cells are more potent than bone marrow-derived cells in a transgenic mouse model of CML, Leukemia, 26, (2012), 1030-1037. |
| [76] | L.O. Ngolet, I. Kocko, A. Elira-Dokekias, Pregnancy and Accelerated Phase of Myeloid Chronic Leukemia Treated with Imatinib: A Case Report from a Developing Country, Case Rep. Hematol., (2016), 6104948. |
| [77] | E. Fimmel, Y. S. Semenov, A. S. Bratus, On optimal and suboptimal treatment strategies for a mathematical model of leukemia, Math. Biosci. Eng., 10 (2013), 151-165. |
| [78] | M. Bocchia, A. Sicuranza, E. Abruzzese, A. Iurlo, S. Sirianni, A. Gozzini, et al., Residual Peripheral Blood CD26+ Leukemic Stem Cells in Chronic Myeloid Leukemia Patients During TKI Therapy and During Treatment-Free Remission, Front. Oncol., 8 (2018), 194 |
| [79] | G. Caocci, B. Martino, M. Greco, E. Abruzzese, M. M. Trawinska, S. Lai, et al., Killer immunoglobulin-like receptors can predict TKI treatment-free remission in chronic myeloid leukemia patients, Exp. Hematol., 43 (2015), 1015-1018. |
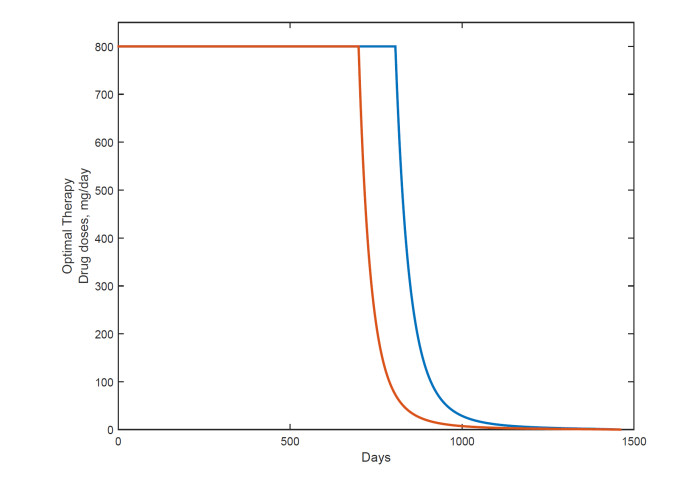
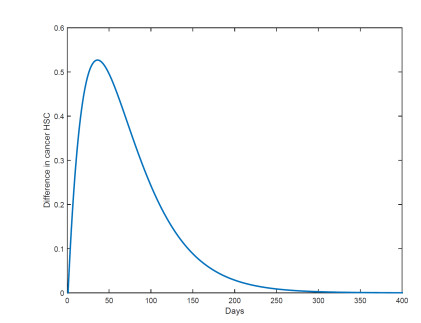
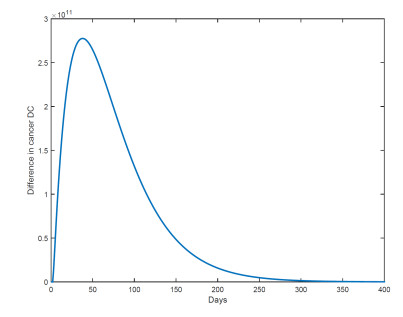
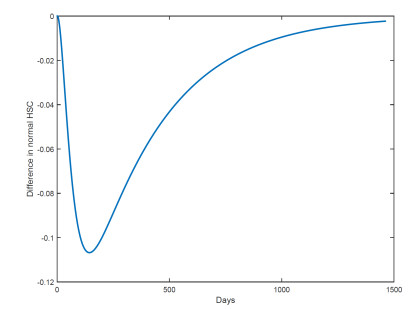
Figures(18) / Tables(1)

Pedro José Gutiérrez-Diez, Jose Russo. Design of personalized cancer treatments by use of optimal control problems: The case of chronic myeloid leukemia[J]. Mathematical Biosciences and Engineering, 2020, 17(5): 4773-4800. doi: 10.3934/mbe.2020261







 DownLoad:
DownLoad: