Citation: Taeyong Lee, Adrianne L. Jenner, Peter S. Kim, Jeehyun Lee. Application of control theory in a delayed-infection and immune-evading oncolytic virotherapy[J]. Mathematical Biosciences and Engineering, 2020, 17(3): 2361-2383. doi: 10.3934/mbe.2020126

| [1] | L.-Q. Fu, S. Wang, M.-H. Cai, X.-J. Wang, J.-Y. Chen, X.-M. Tong, et al., Recent advances in oncolytic virus-based cancer therapy, Virus Res., (2019), 197675. |
| [2] | I. R. Eissa, I. Bustos-Villalobos, T. Ichinose, S. Matsumura, Y. Naoe, N. Miyajima, et al., The current status and future prospects of oncolytic viruses in clinical trials against melanoma, glioma, pancreatic, and breast cancers, Cancers, 10 (2018), 356. |
| [3] | J. Raja, J. M. Ludwig, S. N. Gettinger, K. A. Schalper, H. S. Kim. Oncolytic virus immunotherapy: future prospects for oncology, J. Immunother. Cancer, 6 (2018), 140. |
| [4] | R. Yokoda, B. M. Nagalo, B. Vernon, R. Oklu, H. Albadawi, T. T. DeLeon, et al., Oncolytic virus delivery: from nano-pharmacodynamics to enhanced oncolytic effect, Oncolytic Virotherapy, 6 (2017), 39. |
| [5] | J. W. Choi, E. Kang, O. J. Kwon, T. J. Yun, H. K. Park, P. H. Kim, et al., Local sustained delivery of oncolytic adenovirus with injectable alginate gel for cancer virotherapy, Gene Ther., 20 (2013), 880. |
| [6] | M. A Croyle, N. Chirmule, Y. Zhang, J. M. Wilson. "stealth" adenoviruses blunt cell-mediated and humoral immune responses against the virus and allow for significant gene expression upon readministration in the lung, J. Virol., 75 (2001), 4792-4801. |
| [7] | B.-K. Jung, E. Oh, J. Hong, Y. Lee, K. D. Park, C.-O. Yun, A hydrogel matrix prolongs persistence and promotes specific localization of an oncolytic adenovirus in a tumor by restricting nonspecific shedding and an antiviral immune response, Biomater., 147 (2017), 26-38. |
| [8] | S.-H. Jung, J.-W. Choi, C.-O. Yun, J. Y. Yhee, R. Price, S. H. Kim, et al., Sustained local delivery of oncolytic short hairpin rna adenoviruses for treatment of head and neck cancer, J Gene Med., 16 (2014), 143-152. |
| [9] | E. Oh, J. Oh, J. Hong, Y. Chung, Y. Lee, K. Park, et al., Optimized biodegradable polymeric reservoir-mediated local and sustained co-delivery of dendritic cells and oncolytic adenovirus coexpressing IL-12 and GM-CSF for cancer immunotherapy, J. Control. Release, 259 (2017), 115-127. |
| [10] | J. P. Smith, S. Kanekal, M. B. Patawaran, J. Y. Chen, R. E. Jones, E. K. Orenberg, et al., Drug retention and distribution after intratumoral chemotherapy with fluorouracil/epinephrine injectable gel in human pancreatic cancer xenografts, Cancer Chemoth. Pharm., 44 (1999), 267-274. |
| [11] | S. J. Wade, A. Zuzic, J. Foroughi, S. Talebian, M. Aghmesheh, S. E. Moulton, et al., Preparation and in vitro assessment of wet-spun gemcitabine-loaded polymeric fibers: Towards localized drug delivery for the treatment of pancreatic cancer, Pancreatology, 17 (2017), 795-804. |
| [12] | M. Riley, W. Vermerris, Recent advances in nanomaterials for gene delivery-a review, Nanomaterials, 7 (2017), 94. |
| [13] | J. W. Choi, S. J. Jung, D. Kasala, J. K. Hwang, J. Hu, Y. H. Bae, et al., ph-sensitive oncolytic adenovirus hybrid targeting acidic tumor microenvironment and angiogenesis, J. Control. Release, 205 (2015), 134-143. |
| [14] | S. Tseng, I. M. Kempson, K. Huang, H. Li, Y. Fa, Y. Ho, et al., Targeting tumor microenvironment by bioreduction-activated nanoparticles for light-triggered virotherapy, ACS Nano, 12 (2018), 9894-9902. |
| [15] | A. L. Jenner, F. Frascoli, A. C. F. Coster, P. S. Kim, Enhancing oncolytic virotherapy: Observations from a voronoi cell-based model, J. Theor. Biol., (2019), 110052. |
| [16] | A. L. Jenner, P. S. Kim, F. Frascoli, Oncolytic virotherapy for tumours following a gompertz growth law, J. Theor. Biol., 480 (2019), 129-140. |
| [17] | J. Malinzi, R. Ouifki, A. Eladdadi, D. Torres, K. A. White, Enhancement of chemotherapy using oncolytic virotherapy: mathematical and optimal control analysis, arXiv preprint arXiv:1807.04329, 2018. |
| [18] | E. Ratajczyk, U. Ledzewicz, H. Schättler, Optimal control for a mathematical model of glioma treatment with oncolytic therapy and tnf-α inhibitors, J. Optimiz. Theory App., 176 (2018), 456-477. |
| [19] | M. I. Titze, J. Frank, M. Ehrhardt, S. Smola, N. Graf, T. Lehr, A generic viral dynamic model to systematically characterize the interaction between oncolytic virus kinetics and tumor growth, Eur. J. Pharm. Sci., 97 (2017), 38-46. |
| [20] | T. Cassidy, M. Craig, Determinants of combination gm-csf immunotherapy and oncolytic virotherapy success identified through in silico treatment personalization, PLoS Comput. Biol., 15 (2019), e1007495. |
| [21] | A. Friedman, X. Lai, Combination therapy for cancer with oncolytic virus and checkpoint inhibitor: A mathematical model, PloS One, 13 (2018), e0192449. |
| [22] | W. Mok, T. Stylianopoulos, Y. Boucher, R. K. Jain, Mathematical modeling of herpes simplex virus distribution in solid tumors: implications for cancer gene therapy, Clin. Cancer Res., 15 (2009), 2352-2360. |
| [23] | L. R. Paiva, C. Binny, S. C. Ferreira, M. L. Martins, A multiscale mathematical model for oncolytic virotherapy, Cancer Res., 69 (2009), 1205-1211. |
| [24] | L. G. De Pillis, A. Radunskaya, A mathematical tumor model with immune resistance and drug therapy: an optimal control approach, Comput. Math. Methods Med., 3 (2001), 79-100. |
| [25] | R. Zurakowski, D. Wodarz, Model-driven approaches for in vitro combination therapy using onyx-015 replicating oncolytic adenovirus, J. Theor. Biol., 245 (2007), 1-8. |
| [26] | G. Khan, W. Ahmed, P. S. Philip, M. H. Ali, A. Adem, Healthy rabbits are susceptible to EpsteinBarr virus infection and infected cells proliferate in immunosuppressed animals, Virol. J., 12 (2015), 28. |
| [27] | Z. Z. Wang, Z. M. Guo, H. Smith, A mathematical model of oncolytic virotherapy with time delay, Math. Biosci. Eng., 16 (2019), 1836-1860. |
| [28] | E. Basner-Tschakarjan, F. Mingozzi, Cell-mediated immunity to AAV vectors, evolving concepts and potential solutions, Front. Immun., 5 (2014), 350. |
| [29] | R. H. Byrd, M. E. Hribar, J. Nocedal, An interior point algorithm for large-scale nonlinear programming, SIAM J. Optim., 9 (1999), 877-900. |
| [30] | A. L. Jenner, C.-O. Yun, P. S. Kim, A. C. F. Coster, Mathematical modelling of the interaction between cancer cells and an oncolytic virus: insights into the effects of treatment protocols, Bull. Math. Biol., 80 (2018), 1615-1629. |
| [31] | P. Kim, J. Sohn, J. Choi, Y. Jung, S. Kim, S. Haam, et al., Active targeting and safety profile of peg-modified adenovirus conjugated with herceptin, Biomater., 32 (2011), 2314-2326. |
| [32] | K. Murphy, C. Weaver, Janeway's immunobiology, Garland Science, 2016. |
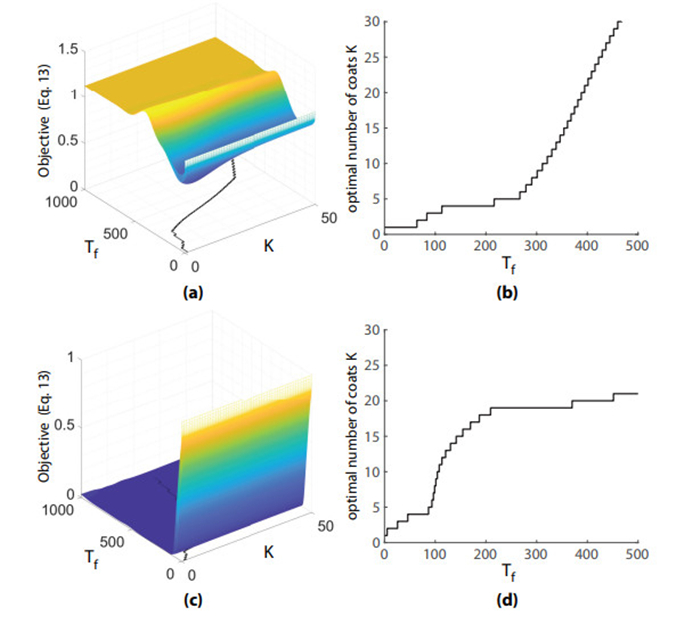
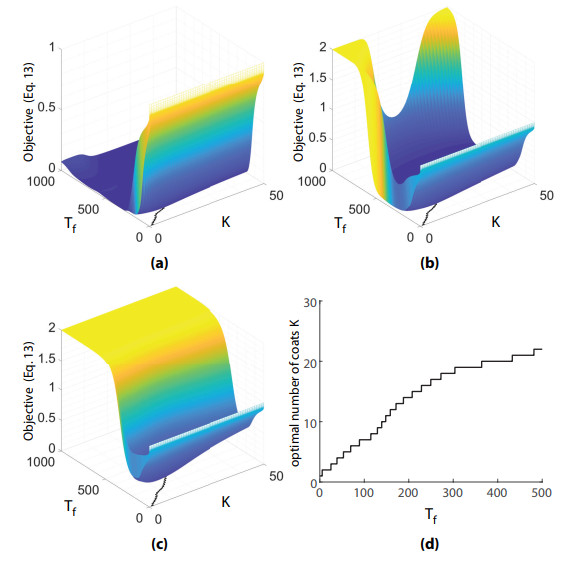
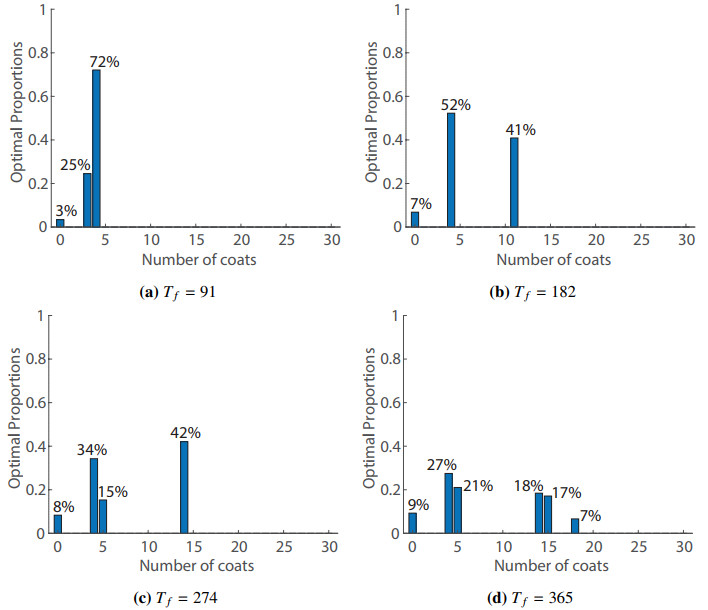
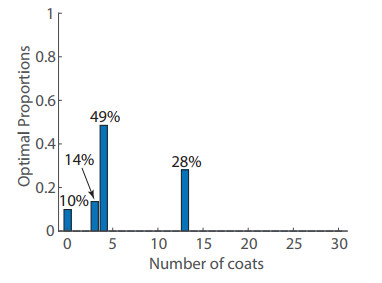
Figures(8) / Tables(1)

Taeyong Lee, Adrianne L. Jenner, Peter S. Kim, Jeehyun Lee. Application of control theory in a delayed-infection and immune-evading oncolytic virotherapy[J]. Mathematical Biosciences and Engineering, 2020, 17(3): 2361-2383. doi: 10.3934/mbe.2020126







 DownLoad:
DownLoad: