Naive human T cells are produced and developed in the thymus, which atrophies abruptly and severely in response to physical or psychological stress. To understand how an instance of stress affects the size and "diversity" of the peripheral naive T cell pool, we derive a mean-field autonomous ODE model of T cell replenishment that allows us to track the clone abundance distribution (the mean number of different TCRs each represented by a specific number of cells). We identify equilibrium solutions that arise at different rates of T cell production, and derive analytic approximations to the dominant eigenvalues and eigenvectors of the mathematical model linearized about these equilibria. From the forms of the eigenvalues and eigenvectors, we estimate rates at which counts of clones of different sizes converge to and depart from equilibrium values-that is, how the number of clones of different sizes "adjusts" to the changing rate of T cell production. Under most physiological realizations of our model, the dominant eigenvalue (representing the slowest dynamics of the clone abundance distribution) scales as a power law in the thymic output for low output levels, but saturates at higher T cell production rates. Our analysis provides a framework for quantitatively understanding how the clone abundance distribution evolves under small changes in the overall T cell production rate.
Citation: Stephanie M. Lewkiewicz, Yao-Li Chuang, Tom Chou. Dynamics of T cell receptor distributions following acute thymic atrophy and resumption[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 28-55. doi: 10.3934/mbe.2020002
Naive human T cells are produced and developed in the thymus, which atrophies abruptly and severely in response to physical or psychological stress. To understand how an instance of stress affects the size and "diversity" of the peripheral naive T cell pool, we derive a mean-field autonomous ODE model of T cell replenishment that allows us to track the clone abundance distribution (the mean number of different TCRs each represented by a specific number of cells). We identify equilibrium solutions that arise at different rates of T cell production, and derive analytic approximations to the dominant eigenvalues and eigenvectors of the mathematical model linearized about these equilibria. From the forms of the eigenvalues and eigenvectors, we estimate rates at which counts of clones of different sizes converge to and depart from equilibrium values-that is, how the number of clones of different sizes "adjusts" to the changing rate of T cell production. Under most physiological realizations of our model, the dominant eigenvalue (representing the slowest dynamics of the clone abundance distribution) scales as a power law in the thymic output for low output levels, but saturates at higher T cell production rates. Our analysis provides a framework for quantitatively understanding how the clone abundance distribution evolves under small changes in the overall T cell production rate.

| [1] | K. Murphy, Immunobiology, Garland Science, 2012. |
| [2] | J. Gameiro, P. Nagib and L. Verinaud, The thymus microenvironment in regulating thymocyte differentiation, Cell Adhes. Migr., 4 (2010), 382–390. |
| [3] | B. Alberts, A. Johnson, J. Lewis, et al., Molecular Biology of the Cell, Garland Science, 2002. |
| [4] | A. Corthday, How do regulatory T cells work?, Scand. J. Immunol., 70 (2009), 326–336. |
| [5] | D. L. Farber, N. A. Yudanin and N. P. Restifo, Human memory T cells: Generation, compartmentalization and homeostasis, Nat. Rev. Immunol., 14 (2014), 24–35. |
| [6] | Y. Takahama, Journey through the thymus: Stromal guides for T-cell development and selection, Nat. Rev. Immunol., 6 (2006), 127–135. |
| [7] | C. H. Bassing, W. Swat and F. W. Alt, The mechanism and regulation of chromosomal V(D)J recombination, Cell, 109 (2002), S45–S55. |
| [8] | D. Mason, A very high level of crossreactivity is an essential feature of the T-cell receptor, Trends Immunol., 19 (1998), 395–404. |
| [9] | J. Nicolić-Žugić, M. K. Slifka and I. Messaoudi, The many important facets of T-cell repertoire diversity, Nat. Rev. Immunol., 4 (2004), 123–132. |
| [10] | D. J. Laydon, C. R. M. Bangham and B. Asquith, Estimating T-cell repertoire diversity: Limitations of classical estimators and a new approach, Philos. Trans. R. Soc. B, 370 (2015), 1–11. |
| [11] | M. S. Chaudhry, E. Velardi, J. A. Dudakov, et al., Thymus: The next (re)generation, Immunol. Rev., 271 (2016), 56–71. |
| [12] | G. G. Steinmann, B. Klaus and H. K. Müller-Hermelink, The involution of the ageing human thymic epithelium is independent of puberty, Scand. J. Immunol., 22 (1985), 563–575. |
| [13] | A. Globerson and R. B. Effros, Aging of lymphocytes and lymphocytes in the aged, Immunol. Today, 21 (2000), 515–521. |
| [14] | A. L. Gruver, L. L. Hudson and J. D. Sempowski, Immunosenescence of aging, J. Pathol., 211 (2007), 144–156. |
| [15] | D. P. Shanley, D. Aw, N. R. Manley, et al., An evolutionary perspective on the mechanisms of immunosenescence, Trends Immunol., 30 (2009), 374–381. |
| [16] | A. L. Gruver and G. D. Sempowski, Cytokines, leptin, and stress-induced thymic atrophy, J. Leukocyte Biol., 84 (2008), 915–923. |
| [17] | J. Dooley and A. Liston. Molecular control over thymic involution: From cytokines and microRNA to aging and adipose tissue, Eur. J. Immunol., 42 (2012), 1073–1079. |
| [18] | H. Selye, Thymus and the adrenals in the response of the organ to injuries and intoxications, Br. J. Exper. Pathol., 17 (1936), 234–248. |
| [19] | W. Savino, The thymus is a common target organ in infectious diseases, PLoS Pathog., 2 (2006), e62. |
| [20] | S. D. Wang, K. J. Huang, Y. S. Lin, et al., Sepsis-induced apoptosis of the thymocytes in mice, J. Immunol., 152 (1994), 5014–5021. |
| [21] | W. W Grody, S. Fliegiel and F. Naeim, Thymus involution in the acquired immunodeficiency syndrome, Am. J. Clin. Pathol., 84 (1985), 85–95. |
| [22] | W. Savino, M. Dardenne, L. A. Velloso, et al., The thymus is a common target in malnutrition and infection, Br. J. Nutr., 98 (2007), S11–S16. |
| [23] | C. L. Mackall, T. A. Fleischer, M. R. Brown, et al., Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy, New Engl. J. Med., 332 (1995), 143–149. |
| [24] | J. Storek, R. P. Witherspoon and R. Storb, T cell reconstitution after bone marrow transplantation into adult patients does not resemble T cell development in early life, Bone Marrow Transplant., 16 (1995), 413–425. |
| [25] | A. L. Zoller, F. J. Schnell and G. J. Kersh, Murine pregnancy leads to reduced proliferation of maternal thymocytes and decreased thymic emigration, Immunology, 121 (2007), 207–215. |
| [26] | T. A. Tibbetts, F. DeMayo, S. Rich, et al., Progesterone receptors in the thymus are required for thymic involution during pregnancy and for normal fertility, PNAS, 96 (1999), 12021–12026. |
| [27] | A. G. Rijhsinghani, K. Thompson and S. K. Bhatia, Estrogen blocks early T cell development in the thymus, Am. J. Reprod. Immunol., 36 (1996), 269–277. |
| [28] | J. D. Ashwell, F. W. M. Lu and M. S. Vacchio, Glucocorticoids in T cell development and function, Annu. Rev. Immunol., 18 (2000), 309–345. |
| [29] | D. A. da Cruz, J. S. Silva, V. C. de Almeida, et al., Altered thymocyte migration during experimental acute trypanosoma cruzi infection: Combined role of fibronectin and the chemokines CXCL12 and CCL4, Eur. J. Immunol., 36 (2006), 1486–1493. |
| [30] | S. K. Stanley, J. M. McCune, H. Kaneshima, et al., Human immunodeficiency virus infection of the human thymus and disruption of the thymic microenvironment in the SCID-hu mouse, J. Exper. Med., 178 (1993), 1151–1163. |
| [31] | M. G. Durdov, O. Springer, V. Ćapkun, et al., The grade of acute thymus involution in neonates correlates with the duration of acute illness and with the percentage of lymphocytes in peripheral blood smear, Biol. Neonate, 83 (2003), 229–234. |
| [32] | J. van Baarlen, H. J. Schuurman, R. Reitsma, et al., Acute thymuc involution during infancy and childhood: Immunohistology of the thymus and peripheral lymphoid tissues after acute illness, Pediat. Pathol., 19 (1989), 261–275. |
| [33] | E. Juretić, A. Juretić, B. Užarević, et al., Alterations in lymphocyte phenotype of preterm infected newborns, Biol. Neonate, 80 (2001), 223–227. |
| [34] | S. M. Falkenberg, C. Johnson, F.V. Bauermann, et al., Changes observed in the thymus and lymph nodes 14 days after exposure to BVDV field strains of enhanced or typical virulence in neonatal calves, Vet. Immunol. Immunopathol., 160 (2014), 70–80. |
| [35] | J. P. L. Rangel, C. S. Galan-Enriquez, M. López-Medina, et al., Bacterial clearance reverses a skewed T-cell repertoire induced by Salmonella infection, Immun., Inflammation Dis., 3 (2015), 209–223. |
| [36] | S. Yovino, L. Kleinberg, S. A. Grossman, et al., The etiology of treatment-related lymphopenia in patients with malignant gliomas: Modeling radiation dose to circulating lymphocytes explains clinical observations and suggests methods of modifying the impact of radiation on immune cells, Cancer Invest., 31 (2013), 140–144. |
| [37] | J. S. Mendez, A. Govindan, J. Leong, et al., Association between treatment-related lymphopenia and overall survival in elderly patients with newly diagnosed glioblastoma, J. Neuro-Oncol., 127 (2016), 329–335. |
| [38] | J. L. Campian, X. Ye, M.Brock, et al., Treatment-related lymphopenia in patients with stage Ⅲ non-small-cell lung cancer, Cancer Invest., 31 (2013), 183–188. |
| [39] | S. S. Long, Laboratory manifestations of infectious disease, in Principles and Practice of Pediatric Infectious Diseases, 4th edition, Elsevier, (2012), 1400–1412. |
| [40] | S. M. Ciupe, B. H. Devlin, M. L. Markert, et al., The dynamics of T-cell receptor repertoire diversity following thymus transplantation for digeorge anomaly, PLoS Comput. Biol., 5 (2009), e1000396. |
| [41] | J. F. Purton, J. A. Monk, D. R. Liddicoat, et al., Expression of the glucocorticoid receptor from the 1A promotor correlates with T lymphocyte sensitivity to glucocorticoid–induced cell death, J. Immunol., 173 (2004), 3816–3824. |
| [42] | F. K. Kong, C. L. H. Chen and M. D. Cooper, Reversible disruption of thymic function by steroid treatment, J. Immunol., 168 (2002), 6500–6505. |
| [43] | J. A. Guevara Patiño, M. W. Marino, V. N. Ivanov, et al., Sex steroids induce apoptosis of CD8+ CD4+ double positive thymocytes via TNF-$\alpha$, Eur. J. Immunol., 30 (2000), 2586–2592. |
| [44] | P. L. Choyke, R. K. Zemon, J. E. Gootenberg, et al., Thymic atrophy and regrowth in response to chemotherapy: CT evaluation, Am. J. Roentgenol., 149 (1987), 269–272. |
| [45] | M. Cohen, C. A. Hill, A. Cangir, et al., Thymic rebound after treatment of childhood tumors, Am. J. Roentgenol., 135 (1980), 151–156. |
| [46] | D. E. DeFriend, J. M. Coote, M. P. Williams, et al., Thymic enlargement in an adult following a severe infection, Clin. Radiol., 56 (2001), 331–333. |
| [47] | D. W. Gelfand, A. S. Goldman, E. J. Law, et al., Thymic hyperplasia in children recovering from thermal burns, J. Trauma, 12 (1972), 813–817. |
| [48] | L. M. Bradley, L. Haynes and S. L. Swain, IL-7: Maintaining T-cell memory and achieving homeostasis, Trends Tmmunol., 26 (2005), 172–176. |
| [49] | J. T. Tan, E. Dudl, E. LeRoy, et al., IL-7 is critical for homeostatic proliferation and survival of naive T cells, Proc. Natl. Acad. Sci., 98 (2001), 8732–8737. |
| [50] | L. Vivien, C. Benoist and D. Mathis, T lymphocytes need IL-7 but not IL-4 or IL-6 to survive in vivo, Int. Immunol., 13 (2001), 763–768. |
| [51] | T. J. Fry and C. L. Mackall, The many faces of IL-7: From lymphopoesis to peripheral T cell maintenance, J. Immunol., 174 (2005), 6571–6576. |
| [52] | S. Xu and T. Chou, Immigration-induced phase transition in a regulated multispecies birth-death process, J. Phys. A: Math. Theor., 51 (2018), 425602. |
| [53] | J. Hataye, J. J. Moon, A. Khoruts, et al., Naïve and memory CD4+ T cell survival controlled by clonal abundance, Science, 312 (2006), 114–116. |
| [54] | R. Dessalles, M. D'Orsogna and T. Chou, Exact steady-state distributions of multispecies birth–death–immigration processes: Effects of mutations and carrying capacity on diversity, J. Stat. Phys., 173 (2018), 182–221. |
| [55] | L. Westera, V. van Hoeven, J. Drylewicz, et al., Lymphocyte maintenance during healthy aging requires no substantial alterations in cellular turnover, Aging Cell, 14 (2015), 219–227. |
| [56] | H. S. Robins, P. V. Campregher, S. K. Srivastava, et al., Comprehensive assessment of T-cell receptor $\beta$-chain diversity in $\alpha \beta$ T cells, Blood, 114 (2009), 4099–4107. |
| [57] | G. Lythe, R. E. Callard, R. L. Hoare, et al., How many TCR clonotypes does a body maintain?, J. Theor. Biol., 389 (2016), 214–224. |
| [58] | S. Lewkiewicz, Y. L. Chuang and T. Chou, A mathematical model of the effects of aging on naive T cell populations and diversity, Bull. Math. Biol., 81 (2019), 2783–2817. |
| [59] | N. Vrisekoop, I. den Braber, A. B. de Boer, et al., Sparse production but preferential incorporation of recently produced naive T-cells in the human peripheral pool, Proc. Natl. Acad. Sci., 105 (2008), 6115–6120. |
| [60] | R. J. de Boer and A. S. Perelson, Quantifying T lymphocyte turnover, J. Theor. Biol., 327 (2013), 45–87. |
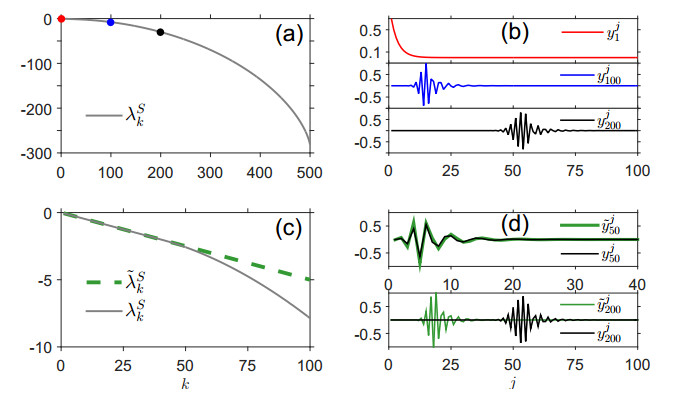
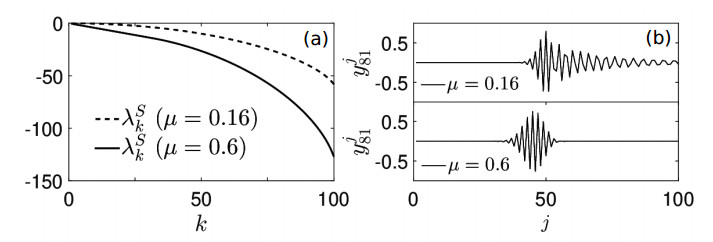
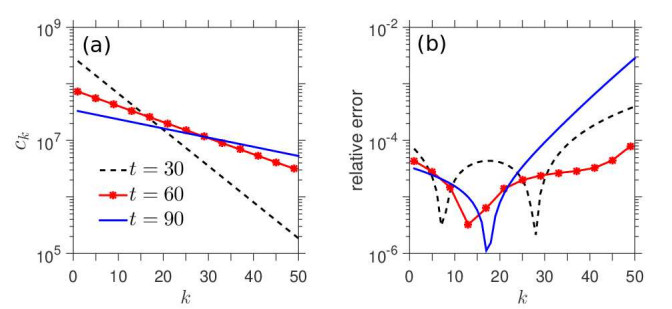
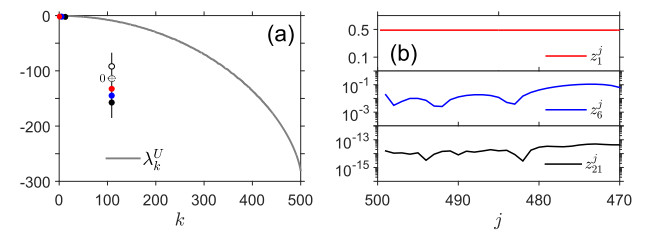
Figures(7)

Stephanie M. Lewkiewicz, Yao-Li Chuang, Tom Chou. Dynamics of T cell receptor distributions following acute thymic atrophy and resumption[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 28-55. doi: 10.3934/mbe.2020002







 DownLoad:
DownLoad: