Citation: Farzad Fatehi, Yuliya N. Kyrychko, Konstantin B. Blyuss. Time-delayed model of autoimmune dynamics[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 5613-5639. doi: 10.3934/mbe.2019279

| [1] | A. K. Abbas, A. H. H. Lichtman and S. Pillai, Cellular and Molecular Immunology, Elsevier Health Sciences, 2015. |
| [2] | D. Mason, A very high level of crossreactivity is an essential feature of the T-cell receptor, Immunol. Today, 19 (1998), 395–404. |
| [3] | E. M. Shevach, R. S. McHugh, C. A. Piccirillo, et al., Control of T-cell activation by CD4+CD25+ suppressor T cells, Immunol. Rev., 182 (2001), 58–67. |
| [4] | A. M. Thornton and E. M. Shevach, CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production, J. Exp. Med., 188 (1998), 287–296. |
| [5] | A. Corthay, How do regulatory T cells work?, Scand. J. Immunol., 70 (2009), 326–336. |
| [6] | D. Buljevac, H. Z. Flach, W. C. J. Hop, et al., Prospective study on the relationship between infections and multiple sclerosis exacerbations, Brain, 125 (2002), 952–960. |
| [7] | D. Germolic, D. H. Kono, J. C. Pfau, et al., Animal models used to examine the role of environment in the development of autoimmune disease: findings from an NIEHS Expert Panel Workshop, J. Autoimmun., 39 (2012), 285–293. |
| [8] | M. P. Mallampalli, E. Davies, D. Wood, et al., Role of environment and sex differences in the development of autoimmune disease: a roundtable meeting report, J. Womens Health, 22 (2013), 578–586. |
| [9] | B. Krone and J. M. Grange, Multiple sclerosis: are protective immune mechanisms compromised by a complex infectious background?, Autoimmune Dis., 2011 (2010), 708750. |
| [10] | M. Ohashi, N. Orlova, C. Quink, et al., Cloning of the Epstein-Barr virus-related rhesus lymphocryptovirus as a bacterial artificial chromosome: a loss-of-function mutation of the rhBARF1 immune evasion gene, J. Virol., 85 (2011), 1330–1339. |
| [11] | D. Hober and P. Sauter, Pathogenesis of type 1 diabetes mellitus: interplay between enterovirus and host, Nat. Rev. Endocrinol., 6 (2010), 279–289. |
| [12] | S. E. Myers, L. Brewer, D. P. Shaw, et al., Prevalent human coxsackie B-5 virus infects porcine islet cells primarily using the coxsackie-adenovirus receptor, Xenotransplantation, 11 (2004), 536–546. |
| [13] | K. Döhner, K. Radtke, S. Schmidt, et al., Eclipse phase of herpes simplex virus type 1 infection: Efficient dynein-mediated capsid transport without the small capsid protein VP26, J. Virol., 80 (2006), 8211–8224. |
| [14] | U. Maurer, B. Sodeik and K. Gruenewald, Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry, Proc. Natl. Acad. Sci. USA, 105 (2008), 10559–10564. |
| [15] | S. Manfredo Vieira, M. Hiltensperger, V. Kumar, et al., Translocation of a gut pathobiont drives autoimmunity in mice and humans, Science, 359 (2018), 1156–1161. |
| [16] | R. S. Fujinami, Can virus infections trigger autoimmune disease?, J. Autoimmun., 16 (2001), 229–234. |
| [17] | A. M. Ercolini and S. D. Miller, The role of infections in autoimmune disease, Clin. Exp. Immunol., 155 (2009), 1–15. |
| [18] | R. S. Fujinami, M. G. von Herrath, U. Christen, et al., Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease, Clin. Microbiol. Rev., 19 (2006), 80–94. |
| [19] | R. S. Fujinami, M. B. Oldstone, Z. Wroblewska, et al., Molecular mimicry in virus infection: crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments, Proc. Natl. Acad. Sci. USA, 80 (1983), 2346–2350. |
| [20] | M. G. von Herrath and M. B. A. Oldstone, Virus-induced autoimmune disease, Curr. Opin. Immunol., 8 (1996), 878–885. |
| [21] | C. Münz, J. D. Lünemann, M. T. Getts, et al., Antiviral immune responses: triggers of or triggered by autoimmunity?, Nat. Rev. Immunol., 9 (2009), 246. |
| [22] | S. Bonhoeffer, R. M. May, G. M. Shaw, et al., Virus dynamics and drug therapy, Proc. Natl. Acad. Sci. USA, 94 (1997), 6971–6976. |
| [23] | M. A. Nowak and C. R. Bangham, Population dynamics of immune responses to persistent viruses, Science-AAAS-Weekly Paper Edition, 272 (1996), 74–79. |
| [24] | A. S. Perelson, Viral kinetics and mathematical models, Am. J. Med., 107 (Suppl 2) (1999), 49–52. |
| [25] | A. S. Perelson, Modelling viral and immune system dynamics, Nat. Rev. Immunol., 2 (2002), 28–36. |
| [26] | P. Baccam, C. Beauchemin, C. A. Macken, et al., Kinetics of influenza A infection in humans, J. Virol., 80 (2006), 7590–7599. |
| [27] | C. A. A. Beauchemin, J. J. McSharry, G. L. Drusano, et al., Modeling amantadine treatment of influenza A virus in vitro, J. Theor. Biol., 254 (2008), 439–451. |
| [28] | C. A. A. Beauchemin and A. Handel, A review of mathematical models of influenza A infections within a host or cell culture: lessons learned and challenges ahead, BMC Public Health, 11 (Suppl 1) (2011), S7. |
| [29] | A. S. Perelson, A. Neumann, M. Markowitz, et al., HIV-1 dynamics in vivo: Virion clearance rate, infected cell life-span, and viral generation time, Science, 271 (1996), 1582–1586. |
| [30] | A. S. Perelson, P. Essunger, Y. Cao, et al., Decay characteristics of HIV-1 infected compartments during combination therapy, Nature, 387 (1997), 188–191. |
| [31] | M. A. Nowak, S. Bonhoeffer, A. M. Hill, et al., Viral dynamics in hepatitis b virus infection, Proc. Natl. Acad. Sci. USA, 93 (1996), 4398–4402. |
| [32] | A. U. Neumann, N. P. Lam, H. Dahari, et al., Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy, Science, 282 (1998), 103–107. |
| [33] | A. U. Neumann, N. P. Lam, H. Dahari, et al., Differences in viral dynamics between genotypes 1 and 2 of hepatitis C virus, J. Infect. Dis., 182 (2000), 28–35. |
| [34] | R. M. Ribeiro, J. Layden-Almer, K. A. Powers, et al., Dynamics of alanine aminotransferase during hepatitis C virus treatment, Hepatology, 38 (2003), 509–517. |
| [35] | L. A. Segel, E. Jäger, D. Elias, et al., A quantitative model of autoimmune disease and T-cell vaccination: does more mean less?, Immunol. Today, 16 (1995), 80–84. |
| [36] | J. A. M. Borghans and R. J. De Boer, A minimal model for T-cell vaccination, Proc. R. Soc. Lond. B Biol. Sci., 259 (1995), 173–178. |
| [37] | J. A. M. Borghans, R. J. De Boer, E. Sercarz, et al., T cell vaccination in experimental autoimmune encephalomyelitis: a mathematical model, J. Immunol., 161 (1998), 1087–1093. |
| [38] | S. Iwami, Y. Takeuchi, Y. Miura, et al., Dynamical properties of autoimmune disease models: tolerance, flare-up, dormancy, J. Theor. Biol., 246 (2007), 646–659. |
| [39] | S. Iwami, Y. Takeuchi, K. Iwamoto, et al., A mathematical design of vector vaccine against autoimmune disease, J. Theor. Biol., 256 (2009), 382–392. |
| [40] | K. León, R. Perez, A. Lage, et al., Modelling T-cell-mediated suppression dependent on interactions in multicellular conjugates, J. Theor. Biol., 207 (2000), 231–254. |
| [41] | K. León, A. Lage and J. Carneiro, Tolerance and immunity in a mathematical model of T-cell mediated suppression, J. Theor. Biol., 225 (2003), 107–126. |
| [42] | K. León, J. Faro, A. Lage, et al., Inverse correlation between the incidences of autoimmune disease and infection predicted by a model of T cell mediated tolerance, J. Autoimmun., 22 (2004), 31–42. |
| [43] | J. Carneiro, T. Paixão, D. Milutinovic, et al., Immunological self-tolerance: lessons from mathematical modeling, J. Comput. Appl. Math., 184 (2005), 77–100. |
| [44] | D. Wodarz and V. A. A. Jansen, A dynamical perspective of CTL cross-priming and regulation: implications for cancer immunology, Immunol. Lett., 86 (2003), 213–227. |
| [45] | R. Root-Bernstein, Theories and modeling of autoimmunity, J. Theor. Biol., 375 (2015), 1–124. |
| [46] | J. D. Fontenot, M. A. Gavin and A. Y. Rudensky, Foxp3 programs the development and function of CD4+CD25+ regulatory T cells, Nat. Immunol., 4 (2003), 330–336. |
| [47] | S. Sakaguchi, Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses, Annu. Rev. Immunol., 22 (2004), 531–562. |
| [48] | S. Z. Josefowicz, L. F. Lu and A. Y. Rudensky, Regulatory T cells: Mechanisms of differentiation and function, Annu. Rev. Immunol., 30 (2012), 531–564. |
| [49] | H. K. Alexander and L. M. Wahl, Self-tolerance and autoimmunity in a regulatory T cell model, Bull. Math. Biol., 73 (2011), 33–71. |
| [50] | N. J. Burroughs, M. Ferreira, B. M. P. M. Oliveira, et al., A transcritical bifurcation in an immune response model, J. Differ. Equ. Appl., 17 (2011), 1101–1106. |
| [51] | N. J. Burroughs, M. Ferreira, B. M. P. M. Oliveira, et al., Autoimmunity arising from bystander proliferation of T cells in an immune response model, Math. Comput. Model., 53 (2011), 1389–1393. |
| [52] | Z. Grossman and W. E. Paul, Adaptive cellular interactions in the immune system: the tunable activation threshold and the significance of subthreshold responses, Proc. Natl. Acad. Sci. USA, 89 (1992), 10365–10369. |
| [53] | Z. Grossman and A. Singer, Tuning of activation thresholds explains flexibility in the selection and development of T cells in the thymus, Proc. Natl. Acad. Sci. USA, 93 (1996), 14747–14752. |
| [54] | Z. Grossman and W. E. Paul, Self-tolerance: context dependent tuning of T cell antigen recognition, Semin. Immunol., 12 (2000), 197–203. |
| [55] | A. J. Noest, Designing lymphocyte functional structure for optimal signal detection: voilá, T cells, J. Theor. Biol., 207 (2000), 195–216. |
| [56] | D. A. Peterson, R. J. DiPaolo, O. Kanagawa, et al., Cutting edge: negative selection of immature thymocytes by a few peptide-MHC complexes: differential sensitivity of immature and mature T cells, J. Immunol., 162 (1999), 3117–3120. |
| [57] | L. B. Nicholson, A. C. Anderson and V. K. Kuchroo, Tuning T cell activation threshold and effector function with cross-reactive peptide ligands, Int. Immunol., 12 (2000), 205–213. |
| [58] | P. Wong, G. M. Barton, K. A. Forbush et al., Dynamic tuning of T cell reactivity by self-peptide-major histocompatibility complex ligands, J. Exp. Med., 193 (2001), 1179–1187. |
| [59] | A. D. Bitmansour, D. C. Douek, V. C. Maino, et al., Direct ex vivo analysis of human CD4+ memory T cell activation requirements at the single clonotype level, J. Immunol., 169 (2002), 1207–1218. |
| [60] | I. Stefanová, J. R. Dorfman and R. N. Germain, Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes, Nature, 420 (2002), 429–434. |
| [61] | G. Altan-Bonnet and R. N. Germain, Modeling T cell antigen discrimination based on feedback control of digital ERK responses, PLoS Biol., 3 (2005), e356. |
| [62] | H. A. van den Berg and D. A. Rand, Dynamics of T cell activation threshold tuning, J. Theor. Biol., 228 (2004), 397–416. |
| [63] | A. Scherer, A. Noest and R. J. de Boer, Activation-threshold tuning in an affinity model for the T-cell repertoire, Proc. R. Soc. Lond. B Biol. Sci., 271 (2004), 609–616. |
| [64] | A. R. McLean, Modelling T cell memory, J. Theor. Biol., 170 (1994), 63–74. |
| [65] | C. Utzny and N. J. Burroughs, Perturbation theory analysis of competition in a heterogeneous population, Physica D, 175 (2003), 109–126. |
| [66] | N. J. Burroughs, B. M. P. M. de Oliveira and A. A. Pinto, Regulatory T cell adjustment of quorum growth thresholds and the control of local immune responses, J. Theor. Biol., 241 (2006), 134–141. |
| [67] | N. J. Burroughs, B. M. P. M. Oliveira, A. A. Pinto, et al., Sensibility of the quorum growth thresholds controlling local immune responses, Math. Comput. Model., 47 (2008), 714–725. |
| [68] | A. L. DeFranco, R. M. Locksley and M. Robertson, Immunity: The immune response in infectious and inflammatory disease, New Science Press Ltd., 2007. |
| [69] | A. Toda and C. A. Piccirillo, Development and function of naturally occurring CD4+CD25+ regulatory T cells, J. Leukoc. Biol., 80 (2006), 458–470. |
| [70] | P. S. Kim, P. P. Lee and D. Levy, Modeling regulation mechanisms in the immune system, J. Theor. Biol., 246 (2007), 33–69. |
| [71] | B. M. P. M. Oliveira, R. Trinchet, M. V. O. Espinar, et al., Modelling the suppression of autoimmunity after pathogen infection, Math. Meth. Appl. Sci., 41 (2018), 8565–8570. |
| [72] | J. Tam, Delay effect in a model for virus replication, IMA J. Math. Appl. Med. Biol., 16 (1999), 29–37. |
| [73] | R. V. Culshaw and S. Ruan, A delay-differential equation model of HIV infection of CD4+ T-cells, Math. Biosci., 165 (2000), 27–39. |
| [74] | P. W. Nelson and A. S. Perelson, Mathematical analysis of delay differential equation models of HIV-1 infection, Math. Biosci., 179 (2002), 73–94. |
| [75] | X. Zhou, X. Song and X. Shi, Analysis of stability and Hopf bifurcation for an HIV infection model with time delay, Appl. Math. Comput., 199 (2008), 23–38. |
| [76] | A. J. Yates, M. Van Baalen and R. Antia, Virus replication strategies and the critical CTL numbers required for the control of infection, PLoS Comput. Biol., 7 (2011), e1002274. |
| [77] | G. J. M. Webster, S. Reignat, M. K. Maini, et al., Incubation phase of acute hepatitis B in man: dynamic of cellular immune mechanisms, Hepatology, 32 (2000), 1117–1124. |
| [78] | M. S. Ciupe, B. L. Bivort, D. M. Bortz, et al., Estimating kinetic parameters from HIV primary infection data through the eyes of three different mathematical models, Math. Biosci., 200 (2006), 1–27. |
| [79] | M. P. Davenport, R. M. Ribeiro and A. S. Perelson, Kinetics of virus-specific CD8+ T cells and the control of human immunodeficiency virus infection, J. Virol., 78 (2004), 10096–10103. |
| [80] | R. Thimme, J. Bukh, H. C. Spangenberg, et al., Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease, Proc. Natl. Acad. Sci. USA, 99 (2002), 15661–15668. |
| [81] | K. B. Blyuss and L. B. Nicholson, The role of tunable activation thresholds in the dynamics of autoimmunity, J. Theor. Biol., 308 (2012), 45–55. |
| [82] | K. B. Blyuss and L. B. Nicholson, Understanding the roles of activation threshold and infections in the dynamics of autoimmune disease, J. Theor. Biol., 375 (2015), 13–20. |
| [83] | D. Ben Ezra and J. V. Forrester, Fundal white dots: the spectrum of a similar pathological process, Br. J. Ophthalmol., 79 (1995), 856–860. |
| [84] | T. F. Davies, D. C. Evered, B. Rees Smith, et al., Value of thyroid-stimulating-antibody determination in predicting the short-term thyrotoxic relapse in Graves' disease, Lancet, 309 (1997), 1181–1182. |
| [85] | A. Nylander and D. A. Hafler, Multiple sclerosis, J. Clin. Invest., 122 (2012), 1180–1188. |
| [86] | F. Fatehi, Y. N. Kyrychko, R. Molchanov, et al., Bifurcations and multi-stability in a model of cytokine-mediated autoimmunity, Int. J. Bif. Chaos, 29 (2019), 1950034. |
| [87] | F. Fatehi, Y. N. Kyrychko and K. B. Blyuss, Effects of viral and cytokine delays on dynamics of autoimmunity, Mathematics, 6 (2018), 66. |
| [88] | F. Fatehi, S. N. Kyrychko, A. Ross, et al., Stochastic effects in autoimmune dynamics, Front. Physiol., 9 (2018), 45. |
| [89] | D. A. Copland, M. S. Wertheim, W. J. Armitage, et al., The clinical time-course of Experimental Autoimmune Uveoretinitis using topical endoscopic fundal imaging with histologic and cellular infiltrate correlation, Invest. Ophthalmol. Vis. Sci., 49 (2008), 5458–5465. |
| [90] | J. Boldison, T. K. Khera, D. A. Copland, et al., A novel pathogenic RBP-3 peptide reveals epitope spreading in persistent experimental autoimmune uveoretinitis, Immunology, 146 (2015), 301–311. |
| [91] | P. Krishnapriya and M. Pitchaimani, Analysis of time delay in viral infection model with immune impairment, J. Appl. Math. Comput., 55 (2017), 421–453. |
| [92] | S. D. Wolf, B. N. Dittel, F. Hardardottir, et al., Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice, J. Exp. Med., 184 (1996), 2271–2278. |
| [93] | H. J. Wu, I. I. Ivanov, J. Darce, et al., Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells, Immunity, 32 (2010), 815–827. |
| [94] | I. Baltcheva, L. Codarri, G. Pantaleo, et al., Lifelong dynamics of human CD4+CD25+ regulatory T cells: Insights from in vivo data and mathematical modeling, J. Theor. Biol., 266 (2010), 307–322. |
| [95] | J. Li, L. Zhang and Z. Wang, Two effective stability criteria for linear time-delay systems with complex coefficients, J. Syst. Sci. Complex., 24 (2011), 835–849. |
| [96] | B. Rahman, K. B. Blyuss and Y. N. Kyrychko, Dynamics of neural systems with discrete and distributed time delays, SIAM J. Appl. Dyn. Syst., 14 (2015), 2069–2095. |
| [97] | A. Skapenko, J. Leipe, P. E. Lipsky, et al., The role of the T cell in autoimmune inflammation, Arthritis Res. Ther., 7(Suppl 2) (2005), S4–S14. |
| [98] | R. Antia, V. V. Ganusov and R. Ahmed, The role of models in understanding CD8+ T-cell memory, Nat. Rev. Immunol., 5 (2005), 101–111. |
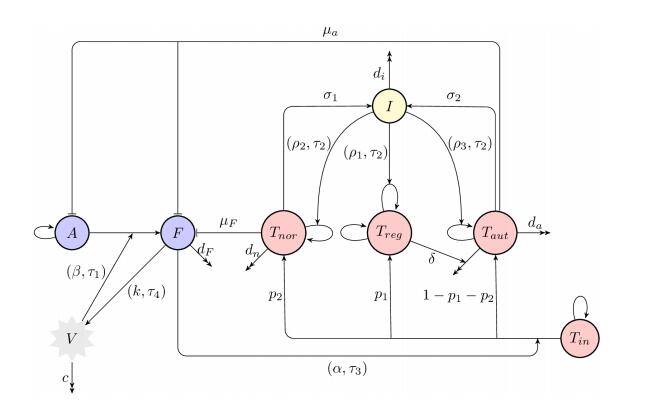
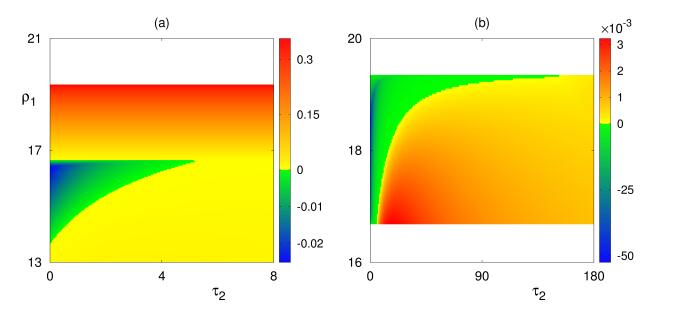
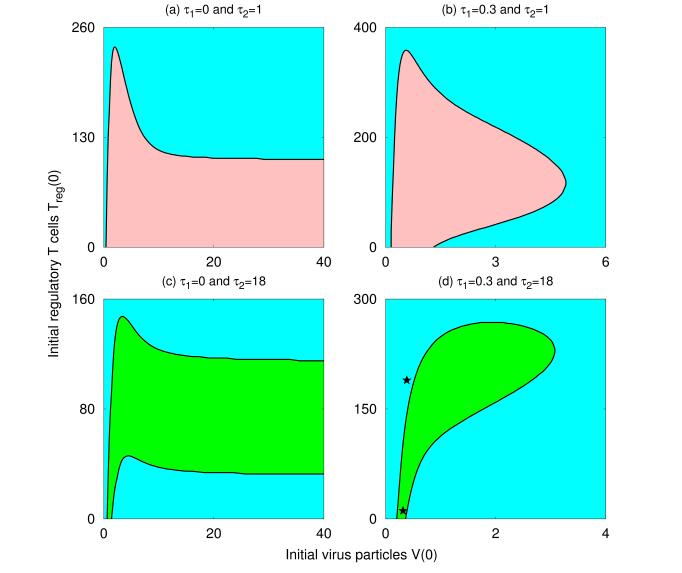
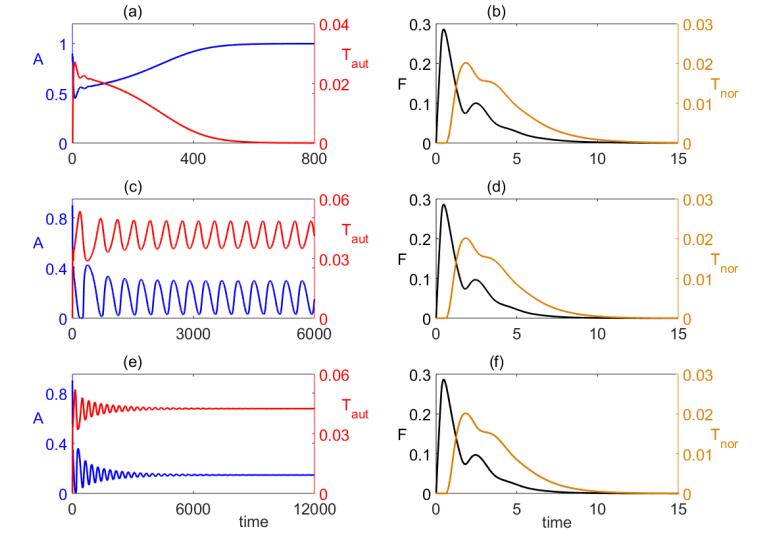
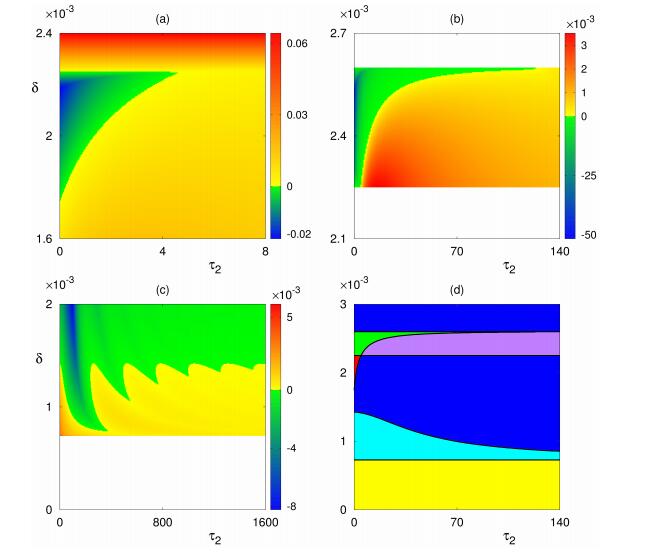
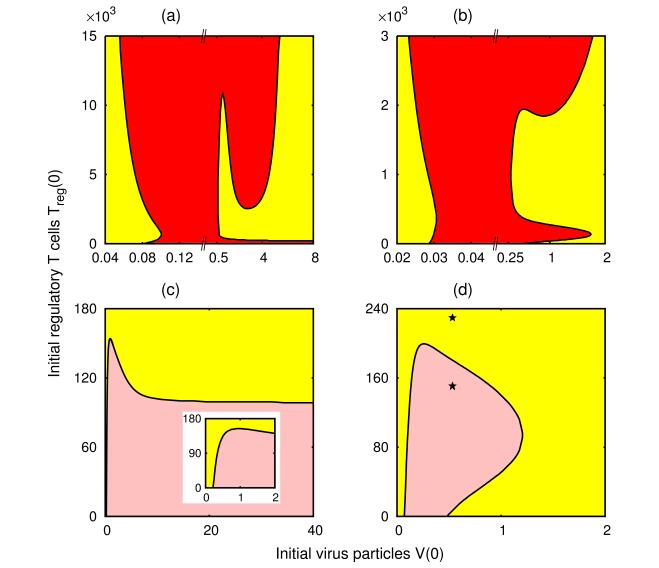
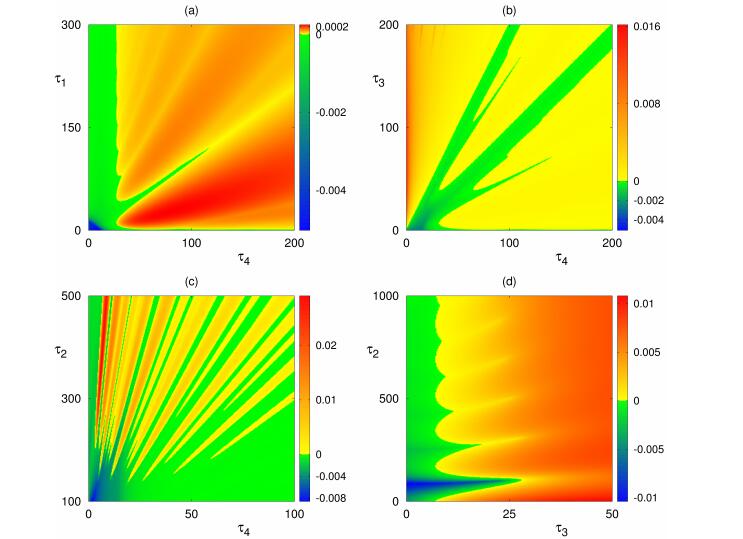
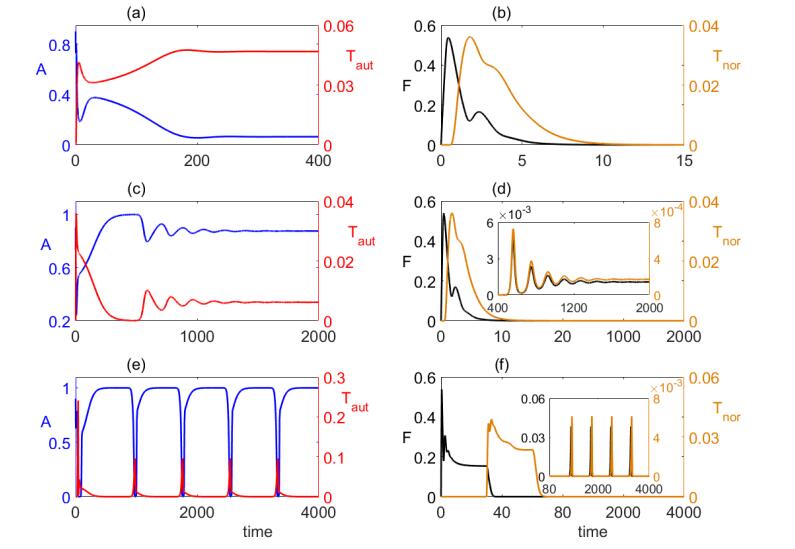
Figures(8) / Tables(1)

Farzad Fatehi, Yuliya N. Kyrychko, Konstantin B. Blyuss. Time-delayed model of autoimmune dynamics[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 5613-5639. doi: 10.3934/mbe.2019279







 DownLoad:
DownLoad: