Citation: Marcella Noorman, Richard Allen, Cynthia J. Musante, H. Thomas Banks. Analysis of compartments-in-series models of liver metabolism as partial differential equations: the effect of dispersion and number of compartments[J]. Mathematical Biosciences and Engineering, 2019, 16(3): 1082-1114. doi: 10.3934/mbe.2019052

| [1] | M. F. Abdelmalek and A. M. Diehl, Nonalcoholic fatty liver disease as a complication of insulin resistance, Med. Clin. North Am., 91 (2007), 1125-1149. |
| [2] | L. A. Adams, P. Angulo and K. D. Lindor, Nonalcoholic fatty liver disease, CMAJ, 172 (2005), 899-905. |
| [3] | C. M. Anderson and A. Stahl, SLC27 fatty acid transport proteins, Mol. Aspects Med., 34 (2013), 516-528. |
| [4] | Y. G. Anissimov and M. S. Roberts, A compartmental model of hepatic disposition kinetics: 1. Model development and application to linear kinetics, J. Pharmacokinet. Pharmacodyn., 29 (2002), 131-156. |
| [5] | J. P. Arab, M. Arrese and M. Trauner, Recent insights into the pathogenesis of nonalcoholic fatty liver disease, Annu. Rev. Pathol. Mech. Dis., 13 (2018), 321-350. |
| [6] | W. B. Ashworth, N. A. Davis and I. D. L. Bogle, A computational model of hepatic energy metabolism: understanding zonated damage and steatosis in NAFLD, PLoS Comput. Biol., 12 (2016), e1005105. |
| [7] | W. B. Ashworth, C. Perez-Galvan, N. A. Davies and I. D. L. Bogle, Liver function as an engineering system, AIChE J., 62 (2016), 3285-3297. |
| [8] | B. A. Banini and A. J. Sanyal, Nonalcoholic fatty liver disease: epidemiology, pathogenesis, natural history, diagnosis, and current treatment options, Clin. Med. Insights Ther., 8 (2016), 75-84. |
| [9] | H. T. Banks, Modeling and Control in the Biomedical Sciences, Lecture Notes in Biomathematics, Vol 6, Springer-Verlag, New York, 1975. |
| [10] | H.T. Banks and H.T. Tran, Mathematical and Experimental Modling of Physical and Biological Processes, Chapman & Hall/CRC Press, Taylor & Francis Group, Boca Raton, 2009. |
| [11] | L. Bass, S. Keiding, K. Winkler and N. Tygstrup, Enzymatic elimination of substrates flowing through the intact liver, J. Theor. Biol., 61 (1976), 393-409. |
| [12] | L. Bass, P. Robinson and A. J. Bracken, Hepatic elimination of flowing substrates: the distributed model, J. Theor. Biol., 72 (1978), 161-184. |
| [13] | N. Berndt, M. S. Horger, S. Bulik and H. G. Holzhütter, A multiscale modelling approach to assess the impact of metabolic zonation and microperfusion on the hepatic carbohydrate metabolism, PLoS Comput. Biol., 14 (2018), e1006005. |
| [14] | S. N. Bhatia, M. Toner, B. D. Foy, A. Rotem, K. M. O'Neil, R. G. Tompkins and M. L. Yarmush, Zonal liver cell heterogeneity: effects of oxygen on metabolic functions of hepatocytes, Cell. Eng., 1 (1996), 125-135. |
| [15] | P. N. Black, C. Ahowesso, D. Montefusco, N. Saini and C. C. DiRusso, Fatty acid transport proteins: targeting FATP2 as a gatekeeper involved in the transport of exogenous fatty acids, Medchemcomm, 7 (2016), 612-622. |
| [16] | P. N. Black, A. Sandoval, E. Arias-Barrau and C. C. DiRusso, Targeting the fatty acid transport proteins (FATP) to understand the mechanisms linking fatty acid transport to metabolism, Immunol. Endocr. Metab. Agents Med. Chem., 9 (2009), 11-17. |
| [17] | E. M. Brunt, Pathology of nonalcoholic fatty liver disease, Nat. Rev. Gastroenterol. Hepatol., 7 (2010), 195-203. |
| [18] | E. M. Brunt, Pathology of fatty liver disease, Mod. Pathol., 20 (2007), S40-S48. |
| [19] | E. M. Brunt, C. G. Janney, A. M. Di Bisceglie, B. A. Neuschwander-Tetri and B. R. Bacon, Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions, Am. J. Gastroenterol., 94 (1999), 2467-2474. |
| [20] | X. Buqué, A. Cano, M. E. Miquilena-Colina, C. García-Monzón, B. Ochoa and P. Aspichueta, High insulin levels are required for FAT/CD36 plasma membrane translocation and enhanced fatty acid uptake in obese Zucker rat hepatocytes, Am. J. Physiol. Endocrinol. Metab., 303 (2012), E504-E514. |
| [21] | D. Calvetti, A. Kuceyeski and E. Somersalo, Sampling-based analysis of a spatially distributed model for liver metabolism at steady state, Multiscale Model. Simul., 7 (2008), 407-431. |
| [22] | G. D. Cartee, Mechanisms for greater insulin-stimulated glucose uptake in normal and insulin-resistant skeletal muscle after acute exercise, Am. J. Physiol. Endocrinol. Metab., 309 (2015), E949-E959. |
| [23] | N. Chalasani, L. Wilson, D. E. Kleiner, O. W. Cummings, E. M. Brunt and A. Ünalp, Relationship of steatosis grade and zonal location to histological features of steatohepatitis in adult patients with non-alcoholic fatty liver disease, J. Hepatol., 48 (2008), 829-834. |
| [24] | E. Chalhoub, L. Xie, V. Balasubramanian, J. Kim and J. Belovich, A distributed model of carbohydrate transport and metabolism in the liver during rest and high-intensity exercise, Ann. Biomed. Eng., 35 (2007), 474-491. |
| [25] | M. Colletti, C. Cicchini, A. Conigliaro, L. Santangelo, T. Alonzi, E. Pasquini, M. Tripodi and L. Amicone, Convergence of Wnt signaling on the HNF4α-driven transcription in controlling liver zonation, Gastroenterology, 137 (2009), 660-672. |
| [26] | S. W. Coppack, R. M. Fisher, G. F. Gibbons, S. M. Humphreys, M. J. McDonough, J. L. Potts and K. N. Frayn, Postprandial substrate deposition in human forearm and adipose tissues in vivo, Clin. Sci. (Lond.), 79 (1990), 339-348. |
| [27] | M. E. Daly, C. Vale, M. Walker, A. Littlefield, K. G. Alberti and J. C. Mathers, Acute effects of insulin sensitivity and diurnal metabolic profiles of a high-sucrose compared with a high-starch diet, Am. J. Clin. Nutr., 67 (1998), 1186-1196. |
| [28] | R. A. DeFronzo and E. Ferrannini, Influence of plasma glucose and insulin concentration on plasma glucose clearance in man, Diabetes, 31 (1982), 683-688. |
| [29] | A. Deussen and J. B. Bassingthwaighte, Modeling [15O] oxygen tracer data for estimating oxygen consumption, Am. J. Physiol., 270 (1996), H1115-H1130. |
| [30] | Gerda de Vries, Thomas Hillen, Mark Lewis, Johannes Muller and Birgitt Schonfisch, A Course in Mathematical Biology: Quantitative Modelling with Mathematical and Computational Methods, SIAM, Philadephia, 2006. |
| [31] | G. Dimitriadis, P. Mitrou, V. Lambadiari, E. Boutati, E. Maratou, E. Koukkou, M. Tzanela, N. Thalassinos and S. A. Raptis, Glucose and lipid fluxes in the adipose tissue after meal ingestion in hyperthyroidism, J. Clin. Endocrinol. Metab., 91 (2006), 1112-1118. |
| [32] | H. Doege, R. A. Baillie, A. M. Ortegon, B. Tsang, Q. Wu, S. Punreddy, D. Hirsch, N. Watson, R. Gimeno and A. Stahl, Targeted deletion of FATP5 reveals multiple functions in liver metabolism: alterations in hepatic lipid homeostasis, Gastroenterology, 130 (2006), 1245-1258. |
| [33] | H. Doege, D. Grimm, A. Falcon, B. Tsang, T. A. Storm, H. Xu, A. M. Ortegon, M. Kazantzis, M. A. Kay and A. Stahl, Silencing of hepatic fatty acid transporter protein 5in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia, J. Biol. Chem., 283 (2008), 22186-22192. |
| [34] | B. Erdogmus, A. Tamer, R. Buyukkaya, B. Yazici, A. Buyukkaya, E. Korkut, A. Alcelik and U. Korkmaz, Portal vein hemodynamics in patients with non-alcoholic fatty liver disease, Tohoku J. Exp. Med., 215 (2008), 89-93. |
| [35] | A. Falcon, H. Doege, A. Fluitt, B. Tsang, N. Watson, M. A. Kay and A. Stahl, FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase, Am. J. Physiol. Endocrinol. Metab., 299 (2010), E384-E393. |
| [36] | S. Fogler, Elements of Chemical Reaction Engineering, 3rd edition, Prentice Hall, New Jersey, 2001. |
| [37] | E. L. Forker and B. Luxon, Hepatic transport kinetics and plasma disappearance curves: distributed modeling vs. conventional approach, Am. J. Physiol., 235 (1978), E648-E660. |
| [38] | R. L. Fournier, Basic Transport Phenomena in Biomedical Engineering, Taylor & Francis, New York, 1998. |
| [39] | X. Fu, J. P. Sluka, S. G. Clendenon1, K. W. Dunn, Z. Wang, J. E. Klaunig and J. A. Glazier, Modeling of Xenobiotic Transport and Metabolism in Virtual Hepatic Lobule Models, PLOS ONE, 13 (2018), e0198060. |
| [40] | R. Gebhardt, Metabolic zonation of the liver: regulation and implications for liver function, Pharmacol. Ther., 53 (1992), 275-354. |
| [41] | Z. Gong, E. Tas, S. Yakar and R. Muzumdar, Hepatic lipid metabolism and non-alcoholic fatty liver disease in aging, Mol. Cell. Endocrinol., 455 (2017), 115-130. |
| [42] | M. R. Gray and Y. K. Tam, The series-compartment model for hepatic elimination, Drug Metab. Dispos., 15 (1987), 27-31. |
| [43] | J. T. Haas, S. Francque and B. Staels, Pathophysiology and mechanisms of nonalcoholic fatty liver disease. Annu. Rev. Physiol., 78 (2016), 181-205. |
| [44] | K. C. Hames, A. Vella, B. J. Kemp and M. D. Jensen, Free fatty acid uptake in humans with CD36 deficiency. Diabetes, 63 (2014), 3606-3614. |
| [45] | T. Hardy, F. Oakley, Q. M. Anstee and C. P. Day, Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu. Rev. Pathol. Mech. Dis., 11 (2016), 451-496. |
| [46] | B. S. Hijmans, A. Grefhorst, M. H. Oosterveer and A. K. Groen, Zonation of glucose and fatty acid metabolism in the liver: mechanism and metabolic consequences, Biochimie, 96 (2014), 121-129. |
| [47] | W. Hundsdorfer and J. G. Verwer, Numerical Solution of Time-Dependent Advection-Diffusion-Reaction Equations, Springer, Berlin, Germany, 2003. |
| [48] | K. Jungermann, Metabolic zonation of liver parenchyma: significance for the regulation of glycogen metabolism, gluconeogenesis, and glycolysis, Diabetes Metab. Rev., 3 (1987), 269-293. |
| [49] | K. Jungermann and N. Katz, Functional hepatocellular heterogeneity, Hepatology, 2 (1982), 385-395. |
| [50] | K. Jungermann and N. Katz, Functional specialization of different hepatocyte populations, Physiol. Rev., 69 (1989), 708-764. |
| [51] | K. Jungermann and T. Kietzmann, Oxygen: modulator of metabolic zonation and disease of the liver, Hepatology, 31 (2000), 255-260. |
| [52] | K. Jungermann and T. Kietzmann, Role of oxygen in the zonation of carbohydrate metabolism and gene expression in liver, Kidney Int., 51 (1997), 402-412. |
| [53] | K. Jungermann and T. Kietzmann, Zonation of parenchymal and nonparenchymal metabolism in liver, Annu. Rev. Nutr., 16 (1996), 179-203. |
| [54] | K. Jungermann and D. Sasse, Heterogeneity of liver parenchymal cells, Trends Biochem. Sci., 3 (1978), P198-P202. |
| [55] | N. R. Katz, Metabolic heterogeneity of hepatocytes across the liver acinus, J. Nutr., 122 (1992), 843-849. |
| [56] | T. Kietzmann, Metabolic zonation of the liver: the oxygen gradient revisited, Redox Biol.,11 (2017), 622-630. |
| [57] | T. Kietzmann, E.Y. Dimova, D. Flúgel and J. G. Scharf, Oxygen: modulator of physiological and pathophysiological processes in the liver, Z. Gastroenterol., 44 (2006), 67-76. |
| [58] | T. Kietzmann and K. Jungermann, Modulation by oxygen of zonal gene expression in liver studied in primary rat hepatocyte cultures, Cell. Biol. Toxicol., 13 (1997), 243-255. |
| [59] | M. Kot, Elements of Mathematical Biology, Cambridge University Press, Cambridge, UK, 2001. |
| [60] | Y. Li, C. C. Chow, A. B. Courville, A. D. Sumner and V. Periwal, Modeling glucose and free fatty acid kinetics in glucose and meal tolerance test, Theor. Biol. Med. Model., 13 (2016). |
| [61] | D. Magalotti, G. Marchesini, S. Ramilli, A. Berzigotti, G. Bianchi and M. Zoli, Splanchnic haemodynamics in non-alcoholic fatty liver disease: effect of a dietary/pharmacological treatment: a pilot study, Dig. Liver Dis., 36 (2004), 406-411. |
| [62] | D. G. Mashek, Hepatic fatty acid trafficking: multiple forks in the road, Adv. Nut., 4 (2013), 697-710. |
| [63] | M. E. Miquilena-Colina, E. Lima-Cabello, S. Sánchez-Campos, M. V. García-Mediavilla, M. Fernández-Bermejo, T. Lozano-Rodríguez, J. Vargas-Castrillón, X. Buqe, B. Ochoa, P. Aspichueta, J. González-Gallego and C. García-Monzón, Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C, Gut, 60 (2011), 1394-1402. |
| [64] | H. Mitsuyoshi, K. Yasui, Y. Harano, M. Endo, K. Tsuji, M. Minami, Y. Itoh, T. Okanoue and T. Yoshikawa, Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease, Hepatol. Res., 39 (2009), 366-373. |
| [65] | A. Mohammadi, M. Ghasemi-rad, H. Zahedi, G. Toldi and T. Alinia, Effect of severity of steatosis as assessed ultrasonographically on hepatic vascular indices in non-alcoholic fatty liver disease, Med. Ultrason., 13 (2011), 200-206. |
| [66] | E. P. Newberry, Y. Xie, S. Kennedy, X. Han, K. K. Buhman, J. Luo, R. W. Gross and N. O. Davidson, Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid-binding protein gene, J. Biol. Chem., 278 (2003), 51664-51672. |
| [67] | H. Ohno, Y. Naito, H. Nakajima and M. Tomita, Construction of a biological tissue model based on a single-cell model: a computer simulation at metabolic heterogeneity in the liver lobule, Artif. Life, 14 (2008), 3-28. |
| [68] | A. Okubo, Difffusion and Ecological Problems: Mathematical Models, Springer-Verlag, Berlin, Heidelberg, New York, 1980. |
| [69] | K. S. Pang, M. Weiss and P. Macheras, Advanced pharmacokinetic models based on organ clearance, circulatory, and fractal concepts, AAPS J., 9 (2007), E268-E283. |
| [70] | S. Park, S. H. J. Kim, G. E. P. Ropella, M. S. Roberts and C. A. Hunt, Tracing multiscale mechanisms of drug disposition in normal and diseased livers, J. Pharmacol. Exp. Ther., 334 (2010), 124-136. |
| [71] | V. Periwal, C. C. Chow, R. N. Bergman, M. Rick, G. L. Vega and A. E. Sumner, Evaluation of quantitative models of the effect of insulin on lipolysis and glucose disposal, Am. J. Physiol. Regul. Integr. Comp. Physiol., 295 (2008), R1089-R1096. |
| [72] | I. Probst, P. Schwartz and K. Jungermann, Induction in primary culture of `gluconeogenic' and `glycolytic' hepatocytes resembling periportal and perivenous cells, Eur. J. Biochem., 126 (1982), 271-278. |
| [73] | G. Rajaraman, M. S. Roberts, D. Hung, G. Q. Wang and F. J. Burczynski, Membrane binding proteins are the major determinants for the hepatocellular transmembrane flux of long-chain fatty acids bound to albumin, Pharm. Res., 22 (2005), 1793-1804. |
| [74] | V Rezania, D. Coombe and J. A. Tuszynski, A physiologically-based flow network model for hepatic drug elimination III: 2D/3D DLA lobule models, Theor. Biol. Med. Model., 13 (2016). |
| [75] | T. Ricken, U. Dahmen and O. Dirsch, A biphasic model for sinusoidal liver perfusion remodeling after outflow obstruction. Biomech. Model. Mechanobiol., 9 (2010), 435-450. |
| [76] | T. Ricken, D. Werner, H. G. Holzhütter, M. König, U. Dahmen and O. Dirsch, Modeling function-perfusion behavior in liver lobules including tissue, blood, glucose, lactate and glycogen by use of a coupled two-scale PDE-ODE approach, Biomech. Model. Mechanobiol., 14 (2015), 515-536. |
| [77] | T. Ricken, N. Waschinsky and D. Werner, Simulation of steatosis zonation in liver lobule-a continuummechanical bi-scale, tri-phasic, multi-component approach, inLecture Notes in Applied and Computational Mechanics, Vol. 84, (eds. P. Wriggers and T. Lenarz), Springer, (2018), 15-33. |
| [78] | M. S. Roberts and M. Rowland, Correlation between in-vitro microsomal enzyme activity and whole organ hepatic elimination kinetics: analysis with a dispersion model, J. Pharm. Pharmacol., 38 (1986), 177-181. |
| [79] | M. S. Roberts and M. Rowland, Hepatic elimination-dispersion model, J. Pharm. Sci., 74 (1985), 585-587. |
| [80] | S. I. Rubinow, Introduction to Mathematical Biology, John Wiley & Sons, New York, 1975. |
| [81] | J. Schleicher, U. Dahmen, R. Guthke and S. Schuster, Zonation of hepatic fat accumulation: insights from mathematical modelling of nutrient gradients and fatty acid uptake, J. R. Soc. Interface, 14 (2017), 20170443. |
| [82] | S. Sheikh-Bahaei, J. J. Maher and C. A. Hunt, Computational experiments reveal plausible mechanisms for changing patterns of hepatic zonation of xenobiotic clearance and hepatotoxicity, J. Theor. Biol., 265 (2010), 718-733. |
| [83] | J. Shi and K. V. Kandror, Study of glucose uptake in adipose cells, Methods Mol. Biol., 456 (2008), 307-315. |
| [84] | J. P. Sluka, X. Fu, Maciej Swat, J. M. Belmonte, A. Cosmanescu, S. G. Clendeno, J. F. Wambaugh, and J. A. Glazier, A liver-centric multiscale modeling framework for xenobiotics, PLOS ONE (11), 2016: DOI:10.1371/journal.pone.0162428. |
| [85] | M. Soresi, L. Giannitrapani, D. Noto, A. Terranova, M. E. Campagna, A. B. Cefal, A. Giammanco and G. Montalto, Effects of steatosis on hepatic hemodynamics in patients with metabolic syndrome, Ultrasound Med. Biol., 41 (2015), 1545-1552. |
| [86] | A. W. Thorburn, B. Gumbiner, F. Bulacan, P. Wallace and R. R. Henry, Intracellular glucose oxidation and glycogen synthase activity are reduced in non-insulin-dependent (type II) diabetes independent of impaired glucose uptake, J. Clin. Invest., 85 (1990), 522-529. |
| [87] | C. Torre, C. Perret and S. Colnot, Molecular determinants of liver zonation, Prog. Mol. Biol. Transl. Sci., 97 (2010), 127-150. |
| [88] | V. van Ginneken, E. de Vries, E. Verheij and J. van der Greef, Potential biomarkers for ``fatty liver'' (hepatic steatosis) and hepatocellular carcinoma (HCC) and an explanation of their pathogenesis, Gastroenterol. Liver Clin. Med., 1 (2017), 001. |
| [89] | D. Wölfle and K. Jungermann, Long-term effects of physiological oxygen concentrations on glycolysis and gluconeogenesis in hepatocyte cultures, Eur. J. Biochem., 151, (1985), 299-303. |
| [90] | Q. Wu, A. M. Ortegon, B. Tsang, H. Doege, K. R. Feingold and A. Stahl, FATP1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity, Mol. Cell. Biol., 26 (2006), 3455-3467. |
| [91] | M. M. Yeh and E. M. Brunt, Pathological features of fatty liver disease, Gastroenterology, 147 (2014), 754-764. |
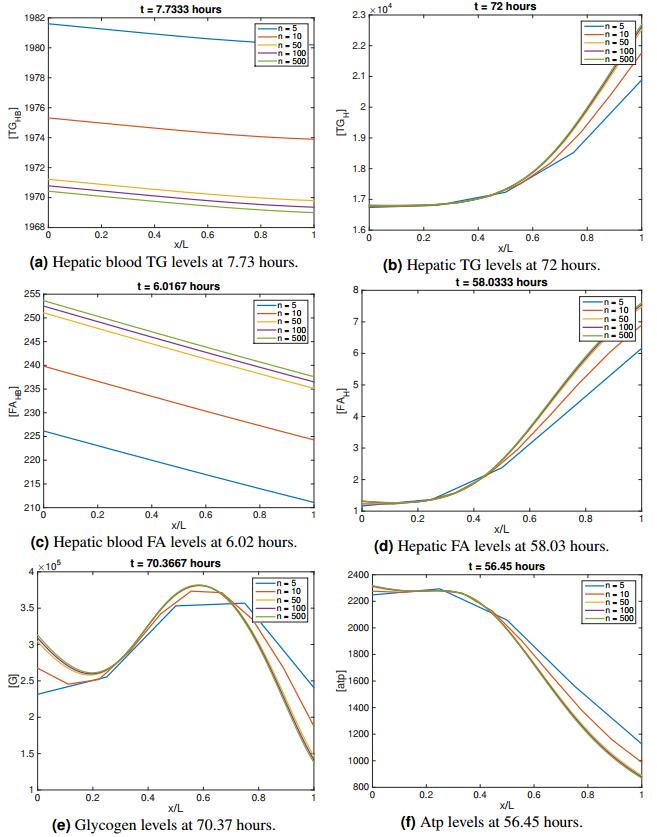
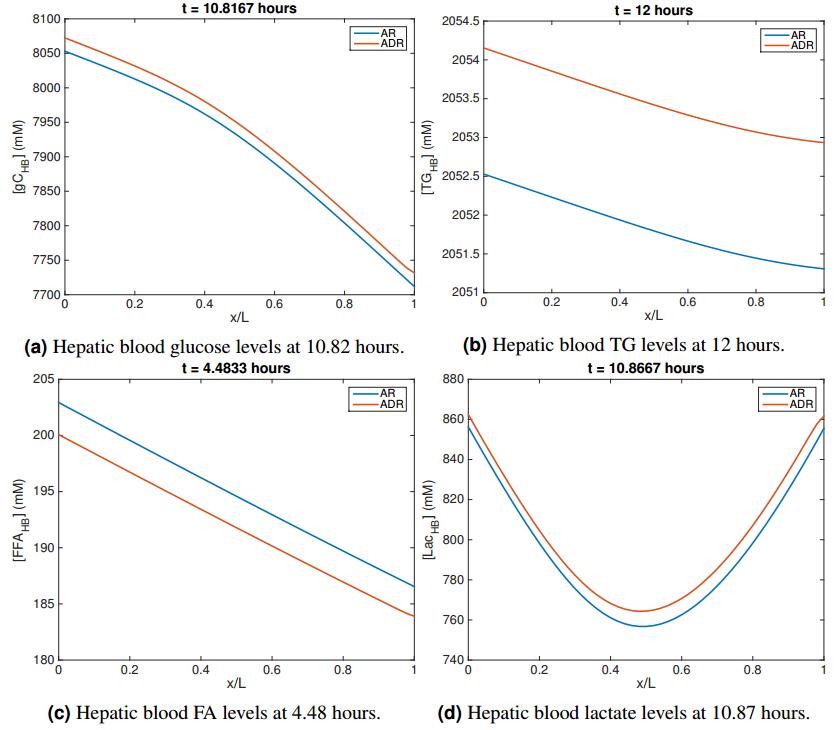
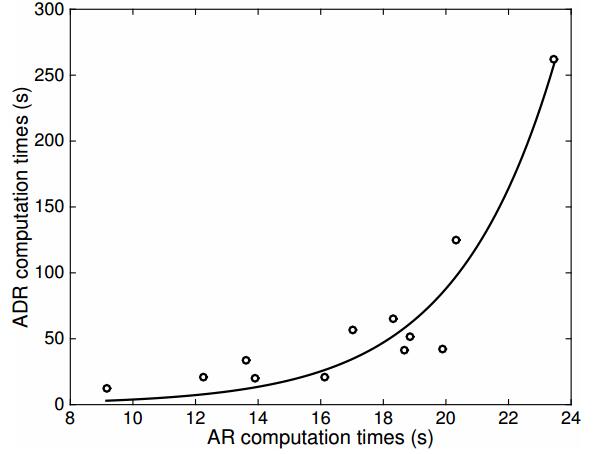
Figures(11) / Tables(11)

Marcella Noorman, Richard Allen, Cynthia J. Musante, H. Thomas Banks. Analysis of compartments-in-series models of liver metabolism as partial differential equations: the effect of dispersion and number of compartments[J]. Mathematical Biosciences and Engineering, 2019, 16(3): 1082-1114. doi: 10.3934/mbe.2019052







 DownLoad:
DownLoad: