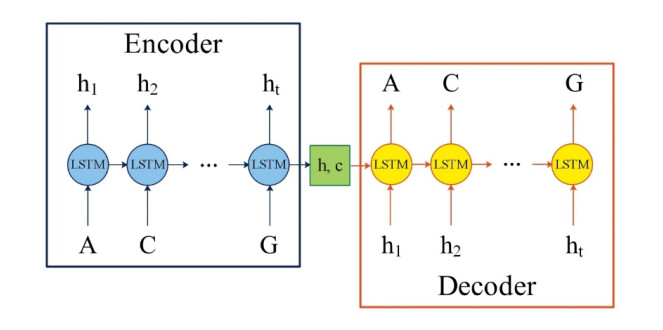
Regulatory elements in DNA sequences, such as promoters, enhancers, terminators and so on, are essential for gene expression in physiological and pathological processes. A promoter is the specific DNA sequence that is located upstream of the coding gene and acts as the "switch" for gene transcriptional regulation. Lots of promoter predictors have been developed for different bacterial species, but only a few are designed for Pseudomonas aeruginosa, a widespread Gram-negative conditional pathogen in nature. In this work, an ensemble model named SPREAD is proposed for the recognition of promoters in Pseudomonas aeruginosa. In SPREAD, the DNA sequence autoencoder model LSTM is employed to extract potential sequence information, and the mean output probability value of CNN and RF is applied as the final prediction. Compared with G4PromFinder, the only state-of-the-art classifier for promoters in Pseudomonas aeruginosa, SPREAD improves the prediction performance significantly, with an accuracy of 0.98, recall of 0.98, precision of 0.98, specificity of 0.97 and F1-score of 0.98.
Citation: Shengming Zhou, Jia Zheng, Cangzhi Jia. SPREAD: An ensemble predictor based on DNA autoencoder framework for discriminating promoters in Pseudomonas aeruginosa[J]. Mathematical Biosciences and Engineering, 2022, 19(12): 13294-13305. doi: 10.3934/mbe.2022622
Regulatory elements in DNA sequences, such as promoters, enhancers, terminators and so on, are essential for gene expression in physiological and pathological processes. A promoter is the specific DNA sequence that is located upstream of the coding gene and acts as the "switch" for gene transcriptional regulation. Lots of promoter predictors have been developed for different bacterial species, but only a few are designed for Pseudomonas aeruginosa, a widespread Gram-negative conditional pathogen in nature. In this work, an ensemble model named SPREAD is proposed for the recognition of promoters in Pseudomonas aeruginosa. In SPREAD, the DNA sequence autoencoder model LSTM is employed to extract potential sequence information, and the mean output probability value of CNN and RF is applied as the final prediction. Compared with G4PromFinder, the only state-of-the-art classifier for promoters in Pseudomonas aeruginosa, SPREAD improves the prediction performance significantly, with an accuracy of 0.98, recall of 0.98, precision of 0.98, specificity of 0.97 and F1-score of 0.98.

| [1] |
N. Masuda, E. Sakagawa, S. Ohya, Outer membrane proteins responsible for multiple drug resistance in Pseudomonas aeruginosa, Antimicrob. Agents Chemother., 39 (1995), 645–649. https://doi.org/10.1128/AAC.39.3.645 doi: 10.1128/AAC.39.3.645

|
| [2] | K. Poole, Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms, J. Mol. Microbiol. Biotechnol., 3 (2001), 255–264. |
| [3] |
G. Bonfiglio, Y. Laksai, L. Franchino, G. Amicosante, G. Nicoletti, Mechanisms of beta-lactam resistance amongst Pseudomonas aeruginosa isolated in an Italian survey, J. Antimicrob. Chemother., 42 (1998), 697–702. https://doi.org/10.1093/jac/42.6.697 doi: 10.1093/jac/42.6.697
|
| [4] |
K. Ohlsen, W. Ziebuhr, K. P. Koller, W. Hell, T. A. Wichelhaus, J. Hacker, Effects of subinhibitory concentrations of antibiotics on alpha-toxin (hla) gene expression of methicillin-sensitive and methicillin-resistant Staphylococcus aureus isolates, Antimicrob. Agents Chemother., 42 (1998), 2817–2823. https://doi.org/10.1128/AAC.42.11.2817 doi: 10.1128/AAC.42.11.2817
|
| [5] |
N. Bagge, O. Ciofu, M. Hentzer, J. I. A. Campbell, M. Givskov, N. Hoiby, Constitutive high expression of chromosomal β-lactamase in Pseudomonas aeruginosa caused by a new insertion sequence (IS 1669) located in ampD, Antimicrob. Agents Chemother., 46 (2002), 3406–3411. https://doi.org/10.1128/AAC.46.11.3406-3411.2002 doi: 10.1128/AAC.46.11.3406-3411.2002
|
| [6] |
P. M. Lepper, E. Grusa, H. Reichl, J. Hogel, M. Trautmann, Consumption of imipenem correlates with β-lactam resistance in Pseudomonas aeruginosa, Antimicrob. Agents Chemother., 46 (2002), 2920–2925. https://doi.org/10.1128/AAC.46.9.2920-2925.2002 doi: 10.1128/AAC.46.9.2920-2925.2002
|
| [7] |
K. J. Hampel, A. E. LaBauve, J. A. Meadows, L. F. Fitzsimmons, A. M. Nock, M. J. Wargo, Characterization of the GbdR regulon in Pseudomonas aeruginosa, J. Bacteriol., 196 (2014), 7–15. https://doi.org/10.1128/JB.01055-13 doi: 10.1128/JB.01055-13
|
| [8] |
L. A. Gallarato, D. G. Sanchez, L. Olvera, E. D. Primo, M. N. Garrido, P. R. Beassoni, et al., Exopolyphosphatase of Pseudomonas aeruginosa is essential for the production of virulence factors, and its expression is controlled by NtrC and PhoB acting at two interspaced promoters, Microbiology-(UK), 160 (2014), 406–417. https://doi.org/10.1099/mic.0.074773-0 doi: 10.1099/mic.0.074773-0
|
| [9] |
W. Liu, Y. Jiang, H. R. Tang, Inferring gene regulatory networks using the improved Markov blanket discovery algorithm, Interdiscip. Sci., 14 (2022), 168–181. https://doi.org/10.1007/s12539-021-00478-9 doi: 10.1007/s12539-021-00478-9
|
| [10] |
R. Amin, C. R. Rahman, S. Ahmed, M. H. R. Sifat, M. N. K. Liton, M. M. Rahman, et al., iPromoter-BnCNN: A novel branched CNN-based predictor for identifying and classifying sigma promoters, Bioinformatics, 36 (2020), 4869–4875. https://doi.org/10.1093/bioinformatics/btaa609 doi: 10.1093/bioinformatics/btaa609
|
| [11] | R. Chevez-Guardado, L. Peña-Castillo, Promotech: A general tool for bacterial promoter recognition, Genome Biol., 22 (2021), 1–16. https://doi.org/10.1186/s13059-021-02514-9 |
| [12] |
A. de Jong, H. Pietersma, M. Cordes, O. P. Kuipers, J. Kok, PePPER: A webserver for prediction of prokaryote promoter elements and regulons, BMC Genomics, 13 (2012), 1–10. https://doi.org/10.1186/1471-2164-13-299 doi: 10.1186/1471-2164-13-299
|
| [13] |
M. D. Salvo, E. Pinatel, A. Tala, M. Fondi, C. Peano, P. Alifano, G4PromFinder: An algorithm for predicting transcription promoters in GC-rich bacterial genomes based on AT-rich elements and G-quadruplex motifs, BMC Bioinf., 19 (2018), 1–11. https://doi.org/10.1186/s12859-018-2049-x doi: 10.1186/s12859-018-2049-x
|
| [14] |
W. Y. He, C. Z. Jia, Y. C. Duan, Q. Zou, 70ProPred: A predictor for discovering sigma70 promoters based on combining multiple features, BMC Syst. Biol., 12 (2018), 99–107. https://doi.org/10.1186/s12918-018-0570-1 doi: 10.1186/s12918-018-0570-1
|
| [15] |
H. Y. Lai, Z. Y. Zhang, Z. D. Su, W. Su, H. Ding, W. Chen, et al., iProEP: A computational predictor for predicting promoter, Mol. Ther. Nucleic Acids, 17 (2019), 337–346. https://doi.org/10.1016/j.omtn.2019.05.028 doi: 10.1016/j.omtn.2019.05.028
|
| [16] |
F. Y. Li, J. X. Chen, Z. Y. Ge, Y. Wen, Y. W. Yue, M. Hayashida, et al., Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework, Briefings Bioinf., 22 (2021), 2126–2140. https://doi.org/10.1093/bib/bbaa049 doi: 10.1093/bib/bbaa049
|
| [17] |
B. Liu, F. Yang, D. S. Huang, K. C. Chou, iPromoter-2L: A two-layer predictor for identifying promoters and their types by multi-window-based PseKNC, Bioinformatics, 34 (2018), 33–40. https://doi.org/10.1093/bioinformatics/btx579 doi: 10.1093/bioinformatics/btx579
|
| [18] |
V. Rangannan, M. Bansal, High-quality annotation of promoter regions for 913 bacterial genomes, Bioinformatics, 26 (2010), 3043–3050. https://doi.org/10.1093/bioinformatics/btq577 doi: 10.1093/bioinformatics/btq577
|
| [19] | V. Salamov, A. Solovyevand, Automatic annotation of microbial genomes and metagenomic sequences, in Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies, (2011), 61–78. |
| [20] |
I. A. Shahmuradov, R. M. Razali, S. Bougouffa, A. Radovanovic, V. B. Bajic, bTSSfinder: A novel tool for the prediction of promoters in cyanobacteria and Escherichia coli, Bioinformatics, 33 (2017), 334–340. https://doi.org/10.1093/bioinformatics/btw629 doi: 10.1093/bioinformatics/btw629
|
| [21] |
R. K. Umarov, V. V. Solovyev, Recognition of prokaryotic and eukaryotic promoters using convolutional deep learning neural networks, PLoS One, 12 (2017), e0171410. https://doi.org/10.1371/journal.pone.0171410 doi: 10.1371/journal.pone.0171410
|
| [22] |
S. Wang, X. S. Cheng, Y. J. Li, M. Wu, Y. H. Zhao, Image-based promoter prediction: A promoter prediction method based on evolutionarily generated patterns, Sci. Rep., 8 (2018), 1–9. https://doi.org/10.1038/s41598-018-36308-0 doi: 10.1038/s41598-018-36308-0
|
| [23] |
M. Zhang, F. Y. Li, T. T. Marquez-Lago, A. Leier, C. Fan, C. K. Kwoh, et al., MULTiPly: A novel multi-layer predictor for discovering general and specific types of promoters, Bioinformatics, 35 (2019), 2957–2965. https://doi.org/10.1093/bioinformatics/btz016 doi: 10.1093/bioinformatics/btz016
|
| [24] |
W. Su, M. L. Liu, Y. H. Yang, J. S. Wang, S. H. Li, H. Lv, et al., PPD: A manually curated database for experimentally verified prokaryotic promoters, J. Mol. Biol., 433 (2021), 166860. https://doi.org/10.1016/j.jmb.2021.166860 doi: 10.1016/j.jmb.2021.166860
|
| [25] |
O. Wurtzel, D. R. Yoder-Himes, K. Han, A. A. Dandekar, S. Edelheit, E. P. Greenberg, et al., The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature, PLoS Pathog., 9 (2012), e1002945. https://doi.org/10.1371/journal.ppat.1002945 doi: 10.1371/journal.ppat.1002945
|
| [26] |
Y. Huang, B. F. Niu, Y. Gao, L. M. Fu, W. Z. Li, CD-HIT Suite: A web server for clustering and comparing biological sequences, Bioinformatics, 26 (2010), 680–682. https://doi.org/10.1093/bioinformatics/btq003 doi: 10.1093/bioinformatics/btq003
|
| [27] |
R. P. Xie, J. H. Li, J. W. Wang, W. Dai, A. Leier, T. T. Marquez-Lago, et al., DeepVF: A deep learning-based hybrid framework for identifying virulence factors using the stacking strategy, Briefings Bioinf., 22 (2021), bbaa125. https://doi.org/10.1093/bib/bbaa125 doi: 10.1093/bib/bbaa125
|
| [28] |
D. D. Zheng, G. S. Pang, B. Liu, L. H. Chen, J. Yang, Learning transferable deep convolutional neural networks for the classification of bacterial virulence factors, Bioinformatics, 36 (2020), 3693–3702. https://doi.org/10.1093/bioinformatics/btaa230 doi: 10.1093/bioinformatics/btaa230
|
| [29] |
R. Mall, A. Elbasir, H. Almeer, Z. Islam, P. R. Kolatkar, S. Chawla, et al., A modeling framework for embedding-based predictions for compound–viral protein activity, Bioinformatics, 37 (2021), 2544–2555. https://doi.org/10.1093/bioinformatics/btab130 doi: 10.1093/bioinformatics/btab130
|
| [30] |
C. C. Wang, C. D. Han, Q. Zhao, X. Chen, Circular RNAs and complex diseases: From experimental results to computational models, Briefings Bioinf., 22 (2021), bbab286. https://doi.org/10.1093/bib/bbab286 doi: 10.1093/bib/bbab286
|
| [31] |
F. Y. Sun, J. Q. Sun, Q, Zhao, A deep learning method for predicting metabolite–disease associations via graph neural network, Briefings Bioinf., 23 (2022), bbac266. https://doi.org/10.1093/bib/bbac266 doi: 10.1093/bib/bbac266
|
| [32] |
Y. Bengio, A. Courville, P. Vincent, Representation learning: A review and new perspectives, IEEE Trans. Pattern Anal. Mach. Intell., 35 (2013), 1798–1828. https://doi.org/10.1109/TPAMI.2013.50 doi: 10.1109/TPAMI.2013.50
|
| [33] |
W. Liu, H. Lin, L. Huang, L. Peng, T. Tang, Q. Zhao, et al., Identification of miRNA-disease associations via deep forest ensemble learning based on autoencoder, Briefings Bioinf., 23 (2022), bbac104. https://doi.org/10.1093/bib/bbac104 doi: 10.1093/bib/bbac104
|
| [34] | U. Michelucci, An introduction to autoencoders, preprint, arXiv: 2201.03898. https://doi.org/10.48550/arXiv.2201.03898 |
| [35] | A. Goyal, A. Lamb, Y. Zhang, S. Z. Zhang, A. Courville, Y. Bengio, Professor forcing: A new algorithm for training recurrent networks, in Advances in Neural Information Processing Systems 29 (NIPS 2016), 29 (2016), 1–9. |
| [36] | T. Q. Chen, C. Guestrin, Xgboost: A scalable tree boosting system, in Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining, (2016), 785–794. https://doi.org/10.1145/2939672.2939785 |
| [37] |
L. Breiman, Random forests, Mach. Learn., 45 (2001), 5–32. https://doi.org/10.1023/A:1010933404324 doi: 10.1023/A:1010933404324
|
| [38] | H. Zhang, The optimality of naive Bayes, Aa, 1 (2004), 3. |
| [39] |
C. Cortes, V. Vapnik, Support-vector networks, Mach. Learn., 20 (1995), 273–297. https://doi.org/10.1007/BF00994018 doi: 10.1007/BF00994018
|
| [40] | J. Laaksonen, E. Oja, Classification with learning k-nearest neighbors, in Proceedings of International Conference on Neural Networks (ICNN'96), 3 (1996), 1480–1483. |
| [41] | Y. LeCun, Y. Bengio, Convolutional networks for images, speech, and time series, Handb. Brain Theory Neural Networks, 3361 (1995), 1995. |
| [42] |
M. Wang, F. Y. Li, H. Wu, Q. Z. Liu, S. Q. Li, PredPromoter-MF (2L): A novel approach of promoter prediction based on multi-source feature fusion and deep forest, Interdiscip. Sci., 14 (2022), 1–15. https://doi.org/10.1007/s12539-022-00520-4 doi: 10.1007/s12539-022-00520-4
|
 mbe-19-12-622-supplementary.docx mbe-19-12-622-supplementary.docx |
 |




Figures(5) / Tables(2)

Shengming Zhou, Jia Zheng, Cangzhi Jia. SPREAD: An ensemble predictor based on DNA autoencoder framework for discriminating promoters in Pseudomonas aeruginosa[J]. Mathematical Biosciences and Engineering, 2022, 19(12): 13294-13305. doi: 10.3934/mbe.2022622







 DownLoad:
DownLoad: