Citation: Christian Costris-Vas, Elissa J. Schwartz, Robert Smith?. Predicting COVID-19 using past pandemics as a guide: how reliable were mathematical models then, and how reliable will they be now?[J]. Mathematical Biosciences and Engineering, 2020, 17(6): 7502-7518. doi: 10.3934/mbe.2020383

| [1] |
Y. Liu, A. A. Gayle, A. Wilder-Smith, J. Rocklöv, The Reproductive Number of COVID-19 Is Higher Compared to SARS Coronavirus, J. Travel Med.e 27 (2020), taaa021. doi: 10.1093/jtm/taaa021

|
| [2] |
Q. Li. X. Guan, P. Wu, X. Wang, L. Zhou, Y. Tong, et al., Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia, N. Engl. J. Med., 382 (2020), 1199-1207. doi: 10.1056/NEJMoa2001316
|
| [3] |
W. Wang, J. Tang, F. Wei, Updated understanding of the outbreak of 2019 novel coronavirus (2019nCoV) in Wuhan, China, J. Med. Virol. 92 (2020), 441-447. doi: 10.1002/jmv.25689
|
| [4] |
K. Kousha, M. Thelwall, COVID-19 publications: Database coverage, citations, readers, tweets, news, Facebook walls, Reddit posts, Quant. Sci. Stud. 1 (2020), 1068-1091. doi: 10.1162/qss_a_00066
|
| [5] |
J. M. Heffernan, R. J. Smith, L. M. Wahl, Perspectives on the basic reproductive ratio, J. R. Soc. Interface 2 (2005), 281-293. doi: 10.1098/rsif.2005.0042
|
| [6] | J. Li, D. Blakeley, R. J. Smith?, The Failure of R0, Comp. Math. Methods Med., 2011 (2011), 527610. |
| [7] |
B. Tang, X. Wang, Q. Li, N. L. Bragazzi, S. Tang, Y. Xiao, et al., Estimation of the Transmission Risk of the 2019-NCoV and Its Implication for Public Health Interventions, J. Clin. Med., 9 (2020), 462. doi: 10.3390/jcm9020462
|
| [8] | N. Imai, A. Cori, I. Dorigatti, M. Baguelin, C. Donnelly, S. Riley, et al., Report 3: Transmissibility of 2019-nCoV, Imperial College London (2020) 1-6. |
| [9] |
S. Zhao, Q. Lin, J. Ran, S. S. Musa, G. Yang, W. Wang, et al., Preliminary estimation of the basic reproduction number of novel coronavirus (2019-nCoV) in China, from 2019 to 2020: A data-driven analysis in the early phase of the outbreak, Int. J. Infect. Dis., 92 (2020), 214-217. doi: 10.1016/j.ijid.2020.01.050
|
| [10] |
J. T. Wu, K. Leung, G.M. Leung, Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study, Lancet, 395 (2020), 689-697. doi: 10.1016/S0140-6736(20)30260-9
|
| [11] | J. Riou, C. L. Althaus, Pattern of early human-to-human transmission of Wuhan 2019-nCoV, Euro. Surveil., 25 (2020), pii = 2000058. |
| [12] |
R. D. Smith, Responding to Global Infectious Disease Outbreaks: Lessons from SARS on the Role of Risk Perception, Communication and Management. Soc. Sci. Med., 63 (2006), 3113-3123. doi: 10.1016/j.socscimed.2006.08.004
|
| [13] | World Health Organization, Summary of Probable SARS Cases with Onset of Illness from 1 November 2002 to 31 July 2003, https://www.who.int/csr/sars/country/table2004_04_21/en/. Accessed 13 Oct 2020. |
| [14] |
S. Riley, C. Fraser, C. A. Donnelly, A. C. Ghani, L. J. Abu-Raddad, A. J. Hedley, et al., Transmission Dynamics of the Etiological Agent of SARS in Hong Kong: Impact of Public Health Interventions, Science, 300 (2003), 1961-1966. doi: 10.1126/science.1086478
|
| [15] |
G. Chowell, P. W. Fenimore, M. A. Castillo-Garsow, C. C. Castillo-Chavez, SARS Outbreaks in Ontario, Hong Kong and Singapore: The Role of Diagnosis and Isolation as a Control Mechanism, J. Theor. Biol., 224 (2003), 1-8. doi: 10.1016/S0022-5193(03)00228-5
|
| [16] |
M. Lipsitch, T. Cohen, B. Cooper, J. M. Robins, S. Ma, L. James, et al., Transmission Dynamics and Control of Severe Acute Respiratory Syndrome, Science, 300 (2003), 1966-1970. doi: 10.1126/science.1086616
|
| [17] | G. Zhou, G. Yan. Severe Acute Respiratory Syndrome Epidemic in Asia, Emerging Infect. Dis., 9 (2003), 1608-1610. |
| [18] |
B. C. K. Choi, A. W. P. Pak. A Simple Approximate Mathematical Model to Predict the Number of Severe Acute Respiratory Syndrome Cases and Deaths, J. Epidemiology Community Health, 57 (2003), 831-835. doi: 10.1136/jech.57.10.831
|
| [19] |
L. O. Lloyd-Smith, A. P. Galvani, W. M. Getz, Curtailing Transmission of Severe Acute Respiratory Syndrome within a Community and Its Hospital, Proc. Royal Soc. B, 270, (2003), 1979-1989. doi: 10.1098/rspb.2003.2481
|
| [20] |
S. A. Eifan, I. Nour, A. Hanif, A. M. Zamzam, S. M. AlJohani, A Pandemic Risk Assessment of Middle East Respiratory Syndrome Coronavirus (MERS-CoV) in Saudi Arabia, Saudi J. Biol. Sci., 24 (2017), 1631-1638. doi: 10.1016/j.sjbs.2017.06.001
|
| [21] |
G. Chowell, F. Abdirizak, S. Lee, J. Lee, E. Jung, H. Nishiura, et al., Transmission Characteristics of MERS and SARS in the Healthcare Setting: A Comparative Study, BMC Med., 13 (2015), 210. doi: 10.1186/s12916-015-0450-0
|
| [22] |
C. Drosten, B. Meyer, M. A. Müller, V. M. Corman, M. Al-Masri, R. Hossain, et al., Transmission of MERS-coronavirus in household contacts, N. Engl. J. Med., 371 (2014), 828-835. doi: 10.1056/NEJMoa1405858
|
| [23] |
S. Cauchemez, C. Fraser, M. D. Van Kerkhove, C. A. Donnelly, S. Riley, A. Rambaut, et al., Middle East Respiratory Syndrome Coronavirus: Quantification of the Extent of the Epidemic, Surveillance Biases, and Transmissibility, Lancet Infect. Dis., 14 (2014), 50-56. doi: 10.1016/S1473-3099(13)70304-9
|
| [24] |
R. Breban, J. Riou, A. Fontanet, Interhuman Transmissibility of Middle East Respiratory Syndrome Coronavirus: Estimation of Pandemic Risk, Lancet, 382 (2013), 694-699. doi: 10.1016/S0140-6736(13)61492-0
|
| [25] |
H.-J. Chang, Estimation of Basic Reproduction Number of the Middle East Respiratory Syndrome Coronavirus (MERS-CoV) during the Outbreak in South Korea, 2015, BioMed. Eng. OnLine, 16 (2017), 79. doi: 10.1186/s12938-017-0370-7
|
| [26] | Centers for Disease Control, H1N1 Flu Pandemic Timeline. Centers for Disease Control and Prevention, https://www.cdc.gov/flu/pandemic-resources/2009-pandemic-timeline.html, Accessed 13 Oct 2020. |
| [27] | T. N. Jilani, R. T. Jamil, A. H. Siddiqui, H1N1 Influenza (Swine Flu). StatPearls, 2020, http://www.ncbi.nlm.nih.gov/books/NBK513241/ Accessed 13 Oct 2020. |
| [28] | Centers for Disease Control, 2009 H1N1 Pandemic (H1N1pdm09 virus), https://www.cdc.gov/flu/pandemic-resources/2009-h1n1-pandemic.html Accessed 13 Oct 2020. |
| [29] |
M. G. Roberts, H. Nishiura, Early Estimation of the Reproduction Number in the Presence of Imported Cases: Pandemic Influenza H1N1-2009 in New Zealand, PLoS ONE, 6 (2011), e17835. doi: 10.1371/journal.pone.0017835
|
| [30] |
C. Fraser, C. A. Donnelly, S. Cauchemez, W. P. Hanage, M. D. Van Kerkhove, T. D. Hollingsworth, et al., Pandemic Potential of a Strain of Influenza A (H1N1): Early Findings, Science, 324 (2009), 1557-1561. doi: 10.1126/science.1176062
|
| [31] | H. Nishiura, C. Castillo-Chavez, M. Safan M, G. Chowell, Transmission Potential of the New Influenza A(H1N1) Virus and Its Age-Specificity in Japan, Euro. Surveil., 14 (2009), 19227. |
| [32] |
L. F. White, J. Wallinga, L. Finelli, C. Reed, S. Riley, M. Lipsitch, et al., Estimation of the Reproductive Number and the Serial Interval in Early Phase of the 2009 Influenza A/H1N1 Pandemic in the USA, Influenza Other Respir. Viruses, 3 (2009), 267-276. doi: 10.1111/j.1750-2659.2009.00106.x
|
| [33] |
L. C. Mostąo-Guidolin, C. S. Bowman, A. L. Greer, D. N. Fisman, S. M. Moghadas, Transmissibility of the 2009 H1N1 Pandemic in Remote and Isolated Canadian Communities: A Modelling Study, BMJ Open, 2 (2012), e001614. doi: 10.1136/bmjopen-2012-001614
|
| [34] |
M. Helferty, J. Vachon, J. Tarasuk, R. Rodin, J. Spika, L. Pelletier, Incidence of Hospital Admissions and Severe Outcomes during the First and Second Waves of Pandemic (H1N1) 2009, CMAJ, 182 (2010), 1981-1987. doi: 10.1503/cmaj.100746
|
| [35] |
O. T. Mytton, P. D. Rutter, M. Mak, E. A. Stanton, N. Sachedina, L. J. Donaldson, Mortality Due to Pandemic (H1N1) 2009 Influenza in England: A Comparison of the First and Second Waves, Epidemiol. Infect., 140 (2012), 1533-1541. doi: 10.1017/S0950268811001968
|
| [36] |
I. Dorigatti, S. Cauchemez, N. M. Ferguson, Increased Transmissibility Explains the Third Wave of Infection by the 2009 H1N1 Pandemic Virus in England, Proc. Natl. Acad. Sci. U.S.A., 110 (2013), 13422-13427. doi: 10.1073/pnas.1303117110
|
| [37] |
O. Sharomi, C. N. Podder, A. B. Gumel, S. M. Mahmud, E. Rubinstein, Modelling the Transmission Dynamics and Control of the Novel 2009 Swine Influenza (H1N1) Pandemic, Bull. Math. Biol., 73 (2011), 515-548. doi: 10.1007/s11538-010-9538-z
|
| [38] |
M. A. Jhung, D. Swerdlow, S. J. Olsen, D. Jernigan, M. Biggerstaff, L. Kamimoto, et al., Epidemiology of 2009 pandemic influenza A (H1N1) in the United States, Clin. Infect. Dis., 52 (2011), S13-S26. doi: 10.1093/cid/ciq008
|
| [39] |
M. D. Van Kerkhove, A. W. Mounts, S. Mall, K. A. Vandemaele, M. Chamberland, T. dos Santos, et al., Epidemiologic and virologic assessment of the 2009 influenza A (H1N1) pandemic on selected temperate countries in the Southern Hemisphere: Argentina, Australia, Chile, New Zealand and South Africa, Influenza Other Respir. Viruses, 5 (2011), e487-e498. doi: 10.1111/j.1750-2659.2011.00249.x
|
| [40] | R. J. Smith?, Did we Eradicate SARS? Lessons Learned and the Way Forward, Am. J. Biomed. Sci. Res., 6 (2019), 001017. |
| [41] |
R. M. Anderson, C. Fraser, A. C. Ghani, C. A. Donnelly, S. Riley, N. M. Ferguson, et al., Epidemiology, Transmission Dynamics and Control of SARS: The 2002-2003 Epidemic, Philos. Trans. R. Soc. Lond., B, Biol. Sci., 359 (2004), 1091-105. doi: 10.1098/rstb.2004.1490
|
| [42] |
S. Ruan, W. Wang, S. A. Levin, The Effect of Global Travel on the Spread of SARS, Math. Biosci. Eng., 3 (2006), 205-218. doi: 10.3934/mbe.2006.3.205
|
| [43] |
N. G. Becker, K. Glass, Z. Li, G. K. Aldis, Controlling Emerging Infectious Diseases like SARS, Math. Biosci., 193 (2005), 205-121. doi: 10.1016/j.mbs.2004.07.006
|
| [44] | A. J. Kucharski, C. L. Althaus, The role of superspreading in Middle East respiratory syndrome coronavirus (MERS-CoV) transmission, Euro. Surveil. 20 (2015), 14-18. |
| [45] |
S. Bernard-Stoecklin, B. Nikolay, A. Assiri, A. A. Saeed, P. K. Embarek, H. El Bushra, et al., Comparative Analysis of Eleven Healthcare-Associated Outbreaks of Middle East Respiratory Syndrome Coronavirus (Mers-Cov) from 2015 to 2017, Sci. Rep., 9 (2019), 1-9. doi: 10.1038/s41598-018-37186-2
|
| [46] |
S. Choi, E. Jung, B. Y. Choi, Y. J. Hur, M. Ki, High Reproduction Number of Middle East Respiratory Syndrome Coronavirus in Nosocomial Outbreaks: Mathematical Modelling in Saudi Arabia and South Korea, J. Hosp. Infect., 99 (2018), 162-168. doi: 10.1016/j.jhin.2017.09.017
|
| [47] |
T. Sardar, I. Ghosh, X. Rodó, J. Chattopadhyay, A Realistic Two-Strain Model for MERS-CoV Infection Uncovers the High Risk for Epidemic Propagation, PLOS Negl. Trop. Dis., 14 (2020), e0008065. doi: 10.1371/journal.pntd.0008065
|
| [48] | M. S. Majumder, C. Rivers, E. Lofgren, D. Fisman. Estimation of MERS-Coronavirus Reproductive Number and Case Fatality Rate for the Spring 2014 Saudi Arabia Outbreak: Insights from Publicly Available Data, PLoS Currents, 6 (2014). |
| [49] |
P. Poletti, M. Ajelli, S. Merler, The effect of risk perception on the 2009 H1N1 pandemic influenza dynamics, PLoS One, 6 (2011), e16460. doi: 10.1371/journal.pone.0016460
|
| [50] | S. Tsukui, Case-Based Surveillance of Pandemic (H1N1) 2009 in Maebashi City, Japan, Jpn. J. Infect. Dis., 65 (2012), 132-137. |
| [51] |
G. Chowell, S. Echevarria-Zuno, C. Viboud, L. Simonsen, J. Tamerius, M. A. Miller, et al., Characterizing the epidemiology of the 2009 influenza A/H1N1 pandemic in Mexico, PLoS Med., 8 (2011), e1000436. doi: 10.1371/journal.pmed.1000436
|
| [52] | C. M. Rivers, E. T. Lofgren, M. Marathe, S. Eubank, B. L. Lewis, Modeling the impact of interventions on an epidemic of Ebola in Sierra Leone and Liberia, PLOS Currents Outbreaks, 6 (2014). |
| [53] | D. Fisman, E. Khoo, A. Tuite, Early epidemic dynamics of the West African 2014 Ebola outbreak: Estimates derived with a simple two-parameter model, PLoS Currents, 6 (2014). |
| [54] |
C. Browne, H. Gulbudak, G. Webb, Modeling contact tracing in outbreaks with application to Ebola, J. Theor. Biol., 384 (2015), 33-49. doi: 10.1016/j.jtbi.2015.08.004
|
| [55] |
G. Webb, C. Browne, A model of the Ebola epidemics in West Africa incorporating age of infection, J. Biol. Dyn., 10 (2016), 18-30. doi: 10.1080/17513758.2015.1090632
|
| [56] |
T. S. Do, Y. S. Lee, Modeling the spread of Ebola. Osong Public Health and Research Perspectives, 7 (2016), 43-48. doi: 10.1016/j.phrp.2015.12.012
|
| [57] | D. Salem, R. Smith?, A Mathematical Model of Ebola Virus Disease: Using Sensitivity Analysis to Determine Effective Intervention Targets, Proceedings of the SummerSim-SCSC 2016 conference, (2016), 16-23. |
| [58] | P. Bhandari, Analysis of Prediction Models in spread of Ebola Virus Disease, Thesis, Deakin University (2019). |
| [59] |
T. C. Germann, K. Kadau, I. M. Longini, C. A. Macken, Mitigation strategies for pandemic influenza in the United States, Proc. Natl. Acad. Sci.U. S.A., 103 (2006), 5935-5940. doi: 10.1073/pnas.0601266103
|
| [60] |
J. T. Wu, B. J. Cowling, The use of mathematical models to inform influenza pandemic preparedness and response, Exp. Biol. Med., 236 (2011), 955-961. doi: 10.1258/ebm.2010.010271
|
| [61] |
S. S. Morse, J. A. Mazet, M. Woolhouse, C. R. Parrish, D. Carroll, W. B. Karesh, et al., Prediction and prevention of the next pandemic zoonosis, Lancet, 380 (2012), 1956-1965. doi: 10.1016/S0140-6736(12)61684-5
|
| [62] |
A. Huppert, G. Katriel, Mathematical modelling and prediction in infectious disease epidemiology, Clin. Microbiol. Infect., 19 (2013), 999-1005. doi: 10.1111/1469-0691.12308
|
| [63] |
P. Saunders-Hastings, B. Q. Hayes, R. Smith? D. Krewski. Modelling community-control strategies to protect hospital resources during an influenza pandemic in Ottawa, Canada, PloS One, 12 (2017), e0179315. doi: 10.1371/journal.pone.0174953
|
| [64] |
M. Valenciano, E. Kissling, J. M. Cohen, N. Oroszi, A. S. Barret, C. Rizzo, et al., Estimates of pandemic influenza vaccine effectiveness in Europe, 2009-2010: results of Influenza Monitoring Vaccine Effectiveness in Europe (I-MOVE) multicentre case-control study, PLoS Med., 8 (2011), e1000388. doi: 10.1371/journal.pmed.1000388
|
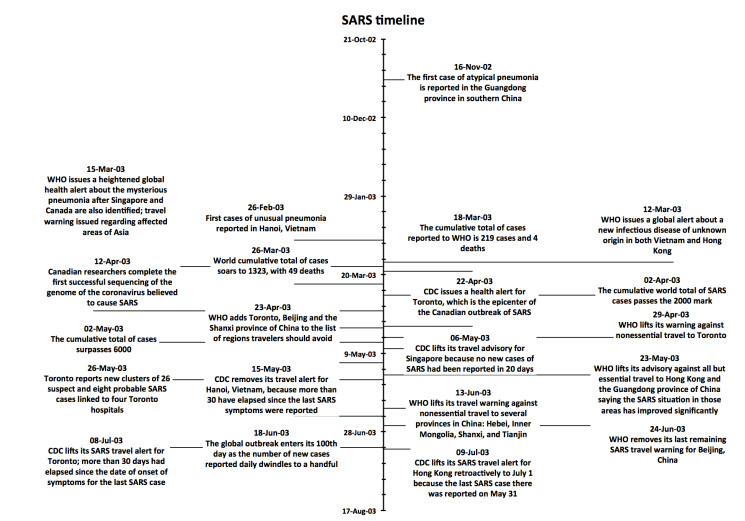
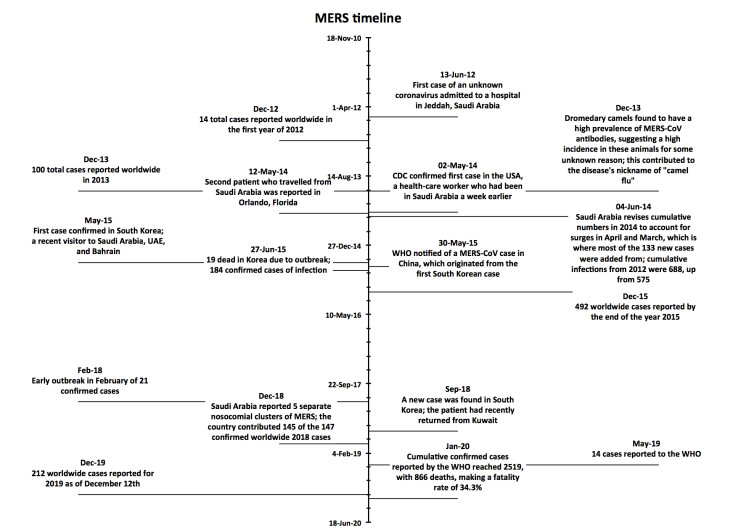
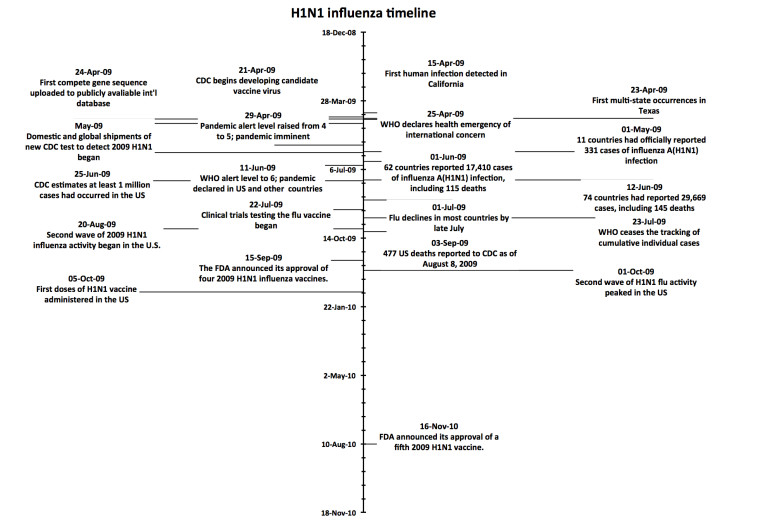
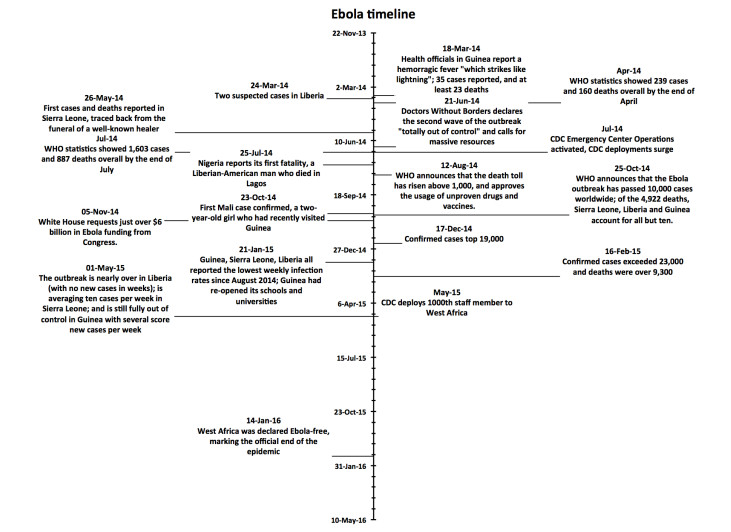
Figures(4)

Christian Costris-Vas, Elissa J. Schwartz, Robert Smith?. Predicting COVID-19 using past pandemics as a guide: how reliable were mathematical models then, and how reliable will they be now?[J]. Mathematical Biosciences and Engineering, 2020, 17(6): 7502-7518. doi: 10.3934/mbe.2020383







 DownLoad:
DownLoad: