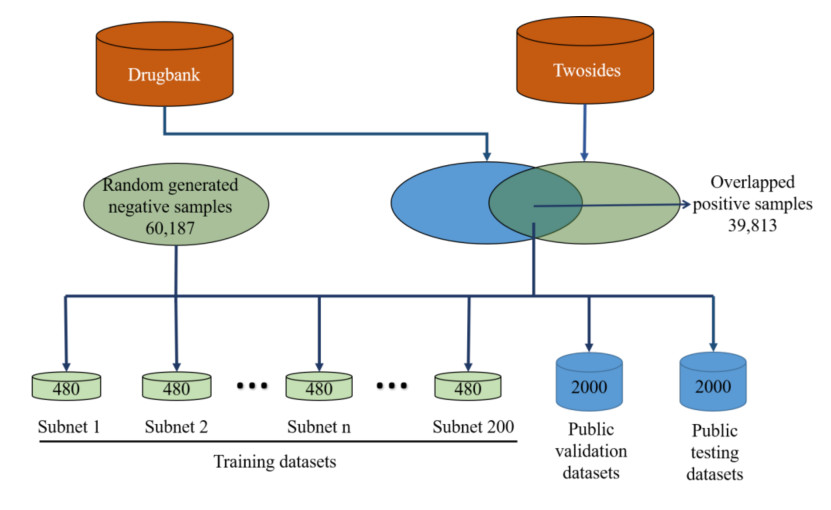
With the increasing need for public health and drug development, combination therapy has become widely used in clinical settings. However, the risk of unanticipated adverse effects and unknown toxicity caused by drug-drug interactions (DDIs) is a serious public health issue for polypharmacy safety. Traditional experimental methods for detecting DDIs are expensive and time-consuming. Therefore, many computational methods have been developed in recent years to predict DDIs with the growing availability of data and advancements in artificial intelligence. In silico methods have proven to be effective in predicting DDIs, but detecting potential interactions, especially for newly discovered drugs without an existing DDI network, remains a challenge. In this study, we propose a predicting method of DDIs named HAG-DDI based on graph attention networks. We consider the differences in mechanisms between DDIs and add learning of semantic-level attention, which can focus on advanced representations of DDIs. By treating interactions as nodes and the presence of the same drug as edges, and constructing small subnetworks during training, we effectively mitigate potential bias issues arising from limited data availability. Our experimental results show that our method achieves an F1-score of 0.952, proving that our model is a viable alternative for DDIs prediction. The codes are available at: https://github.com/xtnenu/DDIFramework.
Citation: Xian Tan, Shijie Fan, Kaiwen Duan, Mengyue Xu, Jingbo Zhang, Pingping Sun, Zhiqiang Ma. A novel drug-drug interactions prediction method based on a graph attention network[J]. Electronic Research Archive, 2023, 31(9): 5632-5648. doi: 10.3934/era.2023286
With the increasing need for public health and drug development, combination therapy has become widely used in clinical settings. However, the risk of unanticipated adverse effects and unknown toxicity caused by drug-drug interactions (DDIs) is a serious public health issue for polypharmacy safety. Traditional experimental methods for detecting DDIs are expensive and time-consuming. Therefore, many computational methods have been developed in recent years to predict DDIs with the growing availability of data and advancements in artificial intelligence. In silico methods have proven to be effective in predicting DDIs, but detecting potential interactions, especially for newly discovered drugs without an existing DDI network, remains a challenge. In this study, we propose a predicting method of DDIs named HAG-DDI based on graph attention networks. We consider the differences in mechanisms between DDIs and add learning of semantic-level attention, which can focus on advanced representations of DDIs. By treating interactions as nodes and the presence of the same drug as edges, and constructing small subnetworks during training, we effectively mitigate potential bias issues arising from limited data availability. Our experimental results show that our method achieves an F1-score of 0.952, proving that our model is a viable alternative for DDIs prediction. The codes are available at: https://github.com/xtnenu/DDIFramework.

| [1] | K. Baxter, Stockley's Drug Interactions : A Source Book of Interactions, Their Mechanisms, Clinical Importance and Management, Pharmaceutica Press, 2010. |
| [2] |
D. M. Qato, J. Wilder, L. P. Schumm, V. Gillet, G. C. Alexander, Changes in prescription and over-the-counter medication and dietary supplement use among older adults in the united states, 2005 vs 2011, JAMA Intern. Med., 176 (2016), 473–482. https://doi.org/10.1001/jamainternmed.2015.8581 doi: 10.1001/jamainternmed.2015.8581

|
| [3] |
Y. Chen, T. Ma, X. Yang, J. Wang, B. Song, X. Zeng, Muffin: Multi-scale feature fusion for drug–drug interaction prediction, Bioinformatics, 37 (2021), 2651–2658. https://doi.org/10.1093/bioinformatics/btab169 doi: 10.1093/bioinformatics/btab169
|
| [4] |
Y. Qiu, Y. Zhang, Y. Deng, S. Liu, W. Zhang, A comprehensive review of computational methods for drug-drug interaction detection, IEEE/ACM Trans. Comput. Biol. Bioinf., 19 (2022), 1968–1985. https://doi.org/10.1109/TCBB.2021.3081268 doi: 10.1109/TCBB.2021.3081268
|
| [5] |
Z. Zhao, Z. Yang, L. Luo, H. Lin, J. Wang, Drug drug interaction extraction from biomedical literature using syntax convolutional neural network, Bioinformatics, 32 (2016), 3444–3453. https://doi.org/10.1093/bioinformatics/btw486 doi: 10.1093/bioinformatics/btw486
|
| [6] | R. Kavuluru, A. Rios, T. Tran, Extracting drug-drug interactions with word and character-level recurrent neural networks, in 2017 IEEE International Conference on Healthcare Informatics, IEEE, (2017), 5–12. |
| [7] |
S. Kim, H. Liu, L. Yeganova, W. J. Wilbur, Extracting drug–drug interactions from literature using a rich feature-based linear kernel approach, J. Biomed. Inf., 55 (2015), 23–30. ttps://doi.org/10.1016/j.jbi.2015.03.002 doi: 10.1016/j.jbi.2015.03.002
|
| [8] | I. N. Dewi, S. Dong, J. Hu, Drug-drug interaction relation extraction with deep convolutional neural networks, in 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), IEEE, (2017), 1795–1802. |
| [9] | Y. Shen, K. Yuan, Y. Li, B. Tang, M. Yang, N. Du, et al., Drug2vec: Knowledge-aware feature-driven method for drug representation learning, in 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), IEEE, (2018), 757–800. |
| [10] |
J. S. Almenoff, W. DuMouchel, L. A. Kindman, X. Yang, D. Fram, Disproportionality analysis using empirical bayes data mining: A tool for the evaluation of drug interactions in the post-marketing setting, Pharmacoepidemiol. Drug Saf., 12 (2003), 517–521. https://doi.org/10.1002/pds.885 doi: 10.1002/pds.885
|
| [11] |
G. N. Norén, A. Bate, R. Orre, I. R. Edwards, Extending the methods used to screen the who drug safety database towards analysis of complex associations and improved accuracy for rare events, Stat. Med., 25 (2006), 3740–3757. https://doi.org/10.1002/sim.2473 doi: 10.1002/sim.2473
|
| [12] |
A. Suzuki, N. Yuen, K. Ilic, R. T. Miller, M. J. Reese, H. R. Brown, et al., Comedications alter drug-induced liver injury reporting frequency: Data mining in the who vigibase™, Regul. Toxicol. Pharm., 72 (2015), 481–490. https://doi.org/10.1001/jamaneurol.2015.0365 doi: 10.1001/jamaneurol.2015.0365
|
| [13] | R. Harpaz, H. S. Chase, C. Friedman, Mining multi-item drug adverse effect associations in spontaneous reporting systems, BMC Bioinf., 11 (2010), 1–8. |
| [14] | Y. Noguchi, A. Ueno, M. Otsubo, H. Katsuno, I. Sugita, Y. Kanematsu, et al., A new search method using association rule mining for drug-drug interaction based on spontaneous report system, Front. Pharmacol., 9 (2018), 197. |
| [15] |
A. Kastrin, P. Ferk, B. Leskošek, Predicting potential drug-drug interactions on topological and semantic similarity features using statistical learning, PLoS One, 13 (2018), e0196865. https://doi.org/10.1371/journal.pone.0196865 doi: 10.1371/journal.pone.0196865
|
| [16] |
C. Yan, G. Duan, Y. Pan, F. X. Wu, J. Wang, Ddigip: Predicting drug-drug interactions based on gaussian interaction profile kernels, BMC Bioinf., 20 (2019), 1–10. https://doi.org/10.1186/s12859-018-2565-8 doi: 10.1186/s12859-018-2565-8
|
| [17] |
S. Qian, S. Liang, H. Yu, Leveraging genetic interactions for adverse drug-drug interaction prediction, PLoS Comput. Biol., 15 (2019), e1007068. https://doi.org/10.1371/journal.pcbi.1007068 doi: 10.1371/journal.pcbi.1007068
|
| [18] | N. Rohani, C. Eslahchi, Drug-drug interaction predicting by neural network using integrated similarity, Sci. Rep., 9 (2019), 13645. |
| [19] | J. Y. Ryu, H. U. Kim, S. Y. Lee, Deep learning improves prediction of drug–drug and drug–food interactions, Proc. Natl. Acad. Sci., 115 (2018), E4304–E4311. |
| [20] |
Y. Deng, X. Xu, Y. Qiu, J. Xia, W. Zhang, S. Liu, A multimodal deep learning framework for predicting drug–drug interaction events, Bioinformatics, 36 (2020), 4316–4322. https://doi.org/10.1093/bioinformatics/btaa501 doi: 10.1093/bioinformatics/btaa501
|
| [21] | S. Liu, Z. Huang, Y. Qiu, Y. P. P. Chen, W. Zhang, Structural network embedding using multi-modal deep auto-encoders for predicting drug-drug interactions, in 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), IEEE, (2019), 445–450. https://doi.org/10.1109/BIBM47256.2019.8983337 |
| [22] |
X. Chen, B. Ren, M. Chen, Q. Wang, L. Zhang, G. Yan, Nllss: Predicting synergistic drug combinations based on semi-supervised learning, PLoS Comput. Biol., 12 (2016), e1004975. https://doi.org/10.1371/journal.pcbi.1004975 doi: 10.1371/journal.pcbi.1004975
|
| [23] | I. Tripodi, K. B. Cohen, L. Hunter, A semantic knowledge-base approach to drug-drug interaction discovery, in 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), IEEE, (2017), 1123–1126. |
| [24] | C. Yan, G. Duan, Y. Zhang, F. X. Wu, Y. Pan, J. Wang, IDNDDI: An integrated drug similarity network method for predicting drug-drug interactions, in Bioinformatics Research and Applications: 15th International Symposium, ISBRA 2019, Springer International Publishing, (2019), 89–99. |
| [25] |
J. Huang, C. Niu, C. D. Green, L. Yang, H. Mei, J. D. J. Han, Systematic prediction of pharmacodynamic drug-drug interactions through protein-protein-interaction network, PLoS Comput. Biol., 9 (2013), e1002998. https://doi.org/10.1371/journal.pcbi.1002998 doi: 10.1371/journal.pcbi.1002998
|
| [26] | R. Karim, M. Cochez, J. Jares, M. Uddin, O. Beyan, S. Decker, Drug-drug interaction prediction based on knowledge graph embeddings and convolutional-lstm network, in Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, ACM, (2019), 113–123. |
| [27] | F. Wang, X. Lei, B. Liao, F. X. Wu, Predicting drug–drug interactions by graph convolutional network with multi-kernel, Briefings Bioinf., 23 (2022), bbab511. |
| [28] | N. Xu, P. Wang, L. Chen, J. Tao, J. Zhao, Mr-gnn: Multi-resolution and dual graph neural network for predicting structured entity interactions, arXiv preprint, (2019), arXiv: 1905.09558. https://doi.org/10.48550/arXiv.1905.09558 |
| [29] |
F. Scarselli, M. Gori, A. C. Tsoi, M. Hagenbuchner, G. Monfardini, The graph neural network model, IEEE Trans. Neural Networks, 20 (2009), 61–80. https://doi.org/10.1109/TNN.2008.2005605 doi: 10.1109/TNN.2008.2005605
|
| [30] | P. Velikovi, G. Cucurull, A. Casanova, A. Romero, P. Liò, Y. Bengio, Graph attention networks, arXiv preprint, (2017), arXiv: 1710.10903. https://doi.org/10.48550/arXiv.1710.10903 |
| [31] | A. K. Nyamabo, H. Yu, J. Y. Shi, Ssi-ddi: Substructure-substructure interactions for drug-drug interaction prediction, Briefings Bioinf., 22 (2021), bbab133. |
| [32] |
Y. H. Feng, S. W. Zhang, Prediction of drug-drug interaction using an attention-based graph neural network on drug molecular graphs, Molecules, 27 (2022), 3004. https://doi.org/10.3390/molecules27093004 doi: 10.3390/molecules27093004
|
| [33] | M. Zitnik, B. Zupan, Collective pairwise classification for multi-way analysis of disease and drug data, in Biocomputing 2016: Proceedings of the Pacific Symposium, (2016), 81–92. |
| [34] | S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, et al., Pubchem in 2021: New data content and improved web interfaces, Nucleic Acids Res., 49 (2021), D1388–D1395. |
| [35] | N. P. Tatonetti, P. P. Ye, R. Daneshjou, R. B. Altman, Data-driven prediction of drug effects and interactions, Sci. Transl. Med., 4 (2012), 125ra31. |
| [36] |
E. Poluzzi, E. Raschi, U. Moretti, F. D. Ponti, Drug-induced torsades de pointes: data mining of the public version of the fda adverse event reporting system (aers), Pharmacoepidemiol. Drug Saf., 18 (2009), 512–518. https://doi.org/10.1016/S0262-1762(09)70172-5 doi: 10.1016/S0262-1762(09)70172-5
|
| [37] |
N. Salim, J. D. Holliday, P. Willett, Combination of fingerprint-based similarity coefficients using data fusion, J. Chem. Inf. Comput. Sci., 43 (2002), 435–442. https://doi.org/10.1021/ci025596j doi: 10.1021/ci025596j
|
| [38] | X. Wang, H. Ji, C. Shi, B. Wang, Y. Ye, P. Cui, et al., Heterogeneous graph attention network, World Wide Web Conf., 2019 (2019), 2022–2032. |
| [39] | B. Perozzi, R. Al-Rfou, S. Skiena, Deepwalk: Online learning of social representations, in Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, ACM, (2014), 701–710. |
| [40] | D. Wang, P. Cui, W. Zhu, Structural deep network embedding, in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, ACM, (2016), 1225–1234. |
| [41] | J. Tang, M. Qu, M. Wang, M. Zhang, J. Yan, Q. Mei, Line: Large-scale information network embedding, in Proceedings of the 24th International Conference on World Wide Web, (2015), 1067–1077. |
| [42] | A. Grover, J. Leskovec, Node2vec: Scalable feature learning for networks, arXiv preprint, (2016), arXiv: 1607.00653. https://doi.org/10.48550/arXiv.1607.00653 |
| [43] | L. Ribeiro, P. Saverese, D. Figueiredo, Struc2vec: Learning node representations from structural identity, in Proceedings of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, ACM, (2017), 385–394. |
| [44] | D. J. Bogaert, L. Verlinden, E. Vandecruys, G. Laureys, E. Verhaeghe, T. Bauters, Severe phototoxicity associated with concomitant use of methotrexate and voriconazole, an overlooked drug-drug interaction, Pediatr. Blood Cancer, 67 (2020), e28246. |
| [45] |
C. J. Parramón-Teixidó, A. Pau-Parra, J. Burgos, D. Campany, Voriconazole and tamsulosin: A clinically relevant drug–drug interaction, Enferm. Infecciosas y Microbiol. Clin., 39 (2021), 361–363. https://doi.org/10.1016/j.eimce.2021.05.005 doi: 10.1016/j.eimce.2021.05.005
|




Figures(5) / Tables(4)
Xian Tan, Shijie Fan, Kaiwen Duan, Mengyue Xu, Jingbo Zhang, Pingping Sun, Zhiqiang Ma. A novel drug-drug interactions prediction method based on a graph attention network[J]. Electronic Research Archive, 2023, 31(9): 5632-5648. doi: 10.3934/era.2023286







 DownLoad:
DownLoad: