Citation: Mariacristina De Luca, Kevin Pels, Susana Moleirinho, Graziella Curtale. The epigenetic landscape of innate immunity[J]. AIMS Molecular Science, 2017, 4(1): 110-139. doi: 10.3934/molsci.2017.1.110

| [1] |
Kimbrell DA, Beutler B (2001) The evolution and genetics of innate immunity. Nat Rev Genet 2: 256-267. doi: 10.1038/35066006

|
| [2] |
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805-820. doi: 10.1016/j.cell.2010.01.022
|
| [3] |
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11: 373-384. doi: 10.1038/ni.1863
|
| [4] |
O'Neill LA, Golenbock D, Bowie AG (2013) The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol 13: 453-460. doi: 10.1038/nri3446
|
| [5] |
Clarke TB (2014) Microbial programming of systemic innate immunity and resistance to infection. PLoS Pathog 10: e1004506. doi: 10.1371/journal.ppat.1004506
|
| [6] |
Drevets DA, Schawang JE, Dillon MJ, et al. (2008) Innate responses to systemic infection by intracellular bacteria trigger recruitment of Ly-6Chigh monocytes to the brain. J Immunol 181: 529-536. doi: 10.4049/jimmunol.181.1.529
|
| [7] | Blach-Olszewska Z, Leszek J (2007) Mechanisms of over-activated innate immune system regulation in autoimmune and neurodegenerative disorders. Neuropsychiatr Dis Treat 3: 365-372. |
| [8] |
Bachmann MF, Kopf M (2001) On the role of the innate immunity in autoimmune disease. J Exp Med 193: F47-50. doi: 10.1084/jem.193.12.F47
|
| [9] | Alvarez-Errico D, Vento-Tormo R, Sieweke M, et al. (2015) Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol 15: 7-17. |
| [10] | Waddington CH (2012) The epigenotype. 1942. Int J Epidemiol 41: 10-13. |
| [11] |
Saeed S, Quintin J, Kerstens HH, et al. (2014) Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345: 1251086. doi: 10.1126/science.1251086
|
| [12] |
Novakovic B, Habibi E, Wang SY, et al. (2016) beta-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 167: 1354-1368. doi: 10.1016/j.cell.2016.09.034
|
| [13] | NE II, Heward JA, Roux B, et al. (2014) Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat Commun 5: 3979. |
| [14] |
Logie C, Stunnenberg HG (2016) Epigenetic memory: A macrophage perspective. Semin Immunol 28: 359-367. doi: 10.1016/j.smim.2016.06.003
|
| [15] |
O'Sullivan TE, Sun JC, Lanier LL (2015) Natural Killer Cell Memory. Immunity 43: 634-645. doi: 10.1016/j.immuni.2015.09.013
|
| [16] |
Netea MG, Quintin J, van der Meer JW (2011) Trained immunity: a memory for innate host defense. Cell Host Microbe 9: 355-361. doi: 10.1016/j.chom.2011.04.006
|
| [17] |
Quintin J, Cheng SC, van der Meer JW, et al. (2014) Innate immune memory: towards a better understanding of host defense mechanisms. Curr Opin Immunol 29: 1-7. doi: 10.1016/j.coi.2014.02.006
|
| [18] |
Netea MG, Joosten LA, Latz E, et al. (2016) Trained immunity: A program of innate immune memory in health and disease. Science 352: aaf1098. doi: 10.1126/science.aaf1098
|
| [19] | Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465-476. |
| [20] | Greer EL, Shi Y (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13: 343-357. |
| [21] |
Fang TC, Schaefer U, Mecklenbrauker I, et al. (2012) Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J Exp Med 209: 661-669. doi: 10.1084/jem.20112343
|
| [22] |
Martinez P, Denys A, Delos M, et al. (2015) Macrophage polarization alters the expression and sulfation pattern of glycosaminoglycans. Glycobiology 25: 502-513. doi: 10.1093/glycob/cwu137
|
| [23] |
Loke P, Nair MG, Parkinson J, et al. (2002) IL-4 dependent alternatively-activated macrophages have a distinctive in vivo gene expression phenotype. BMC Immunol 3: 7. doi: 10.1186/1471-2172-3-7
|
| [24] |
Jenkins SJ, Ruckerl D, Thomas GD, et al. (2013) IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J Exp Med 210: 2477-2491. doi: 10.1084/jem.20121999
|
| [25] |
Cabanel M, Brand C, Oliveira-Nunes MC, et al. (2015) Epigenetic Control of Macrophage Shape Transition towards an Atypical Elongated Phenotype by Histone Deacetylase Activity. PLoS One 10: e0132984. doi: 10.1371/journal.pone.0132984
|
| [26] |
Yang X, Wang X, Liu D, et al. (2014) Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol Endocrinol 28: 565-574. doi: 10.1210/me.2013-1293
|
| [27] |
Ramirez-Carrozzi VR, Nazarian AA, Li CC, et al. (2006) Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev 20: 282-296. doi: 10.1101/gad.1383206
|
| [28] |
Ramirez-Carrozzi VR, Braas D, Bhatt DM, et al. (2009) A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138: 114-128. doi: 10.1016/j.cell.2009.04.020
|
| [29] |
Satoh T, Takeuchi O, Vandenbon A, et al. (2010) The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 11: 936-944. doi: 10.1038/ni.1920
|
| [30] |
Stender JD, Glass CK (2013) Epigenomic control of the innate immune response. Curr Opin Pharmacol 13: 582-587. doi: 10.1016/j.coph.2013.06.002
|
| [31] | Blackwood EM, Kadonaga JT (1998) Going the distance: a current view of enhancer action. Science 281: 60-63. |
| [32] |
Kaikkonen MU, Spann NJ, Heinz S, et al. (2013) Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell 51: 310-325. doi: 10.1016/j.molcel.2013.07.010
|
| [33] | Pott S, Lieb JD (2015) What are super-enhancers? Nat Genet 47: 8-12. |
| [34] |
Brown JD, Lin CY, Duan Q, et al. (2014) NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell 56: 219-231. doi: 10.1016/j.molcel.2014.08.024
|
| [35] |
Price AE, Liang HE, Sullivan BM, et al. (2010) Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A 107: 11489-11494. doi: 10.1073/pnas.1003988107
|
| [36] |
Monticelli LA, Sonnenberg GF, Abt MC, et al. (2011) Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12: 1045-1054. doi: 10.1038/ni.2131
|
| [37] |
Fuchs A, Vermi W, Lee JS, et al. (2013) Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 38: 769-781. doi: 10.1016/j.immuni.2013.02.010
|
| [38] |
Cella M, Fuchs A, Vermi W, et al. (2009) A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 457: 722-725. doi: 10.1038/nature07537
|
| [39] |
Buonocore S, Ahern PP, Uhlig HH, et al. (2010) Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464: 1371-1375. doi: 10.1038/nature08949
|
| [40] |
Goto Y, Ivanov, II (2013) Intestinal epithelial cells as mediators of the commensal-host immune crosstalk. Immunol Cell Biol 91: 204-214. doi: 10.1038/icb.2012.80
|
| [41] |
Salzman NH, Underwood MA, Bevins CL (2007) Paneth cells, defensins, and the commensal microbiota: a hypothesis on intimate interplay at the intestinal mucosa. Semin Immunol 19: 70-83. doi: 10.1016/j.smim.2007.04.002
|
| [42] |
Fischer N, Sechet E, Friedman R, et al. (2016) Histone deacetylase inhibition enhances antimicrobial peptide but not inflammatory cytokine expression upon bacterial challenge. Proc Natl Acad Sci U S A 113: E2993-3001. doi: 10.1073/pnas.1605997113
|
| [43] |
Chookajorn T, Dzikowski R, Frank M, et al. (2007) Epigenetic memory at malaria virulence genes. Proc Natl Acad Sci U S A 104: 899-902. doi: 10.1073/pnas.0609084103
|
| [44] |
Huguenin M, Bracha R, Chookajorn T, et al. (2010) Epigenetic transcriptional gene silencing in Entamoeba histolytica: insight into histone and chromatin modifications. Parasitology 137: 619-627. doi: 10.1017/S0031182009991363
|
| [45] |
Marazzi I, Ho JS, Kim J, et al. (2012) Suppression of the antiviral response by an influenza histone mimic. Nature 483: 428-433. doi: 10.1038/nature10892
|
| [46] |
Pennini ME, Pai RK, Schultz DC, et al. (2006) Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-gamma-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol 176: 4323-4330. doi: 10.4049/jimmunol.176.7.4323
|
| [47] | Lebreton A, Job V, Ragon M, et al. (2014) Structural basis for the inhibition of the chromatin repressor BAHD1 by the bacterial nucleomodulin LntA. MBio 5: e00775-00713. |
| [48] |
Eskandarian HA, Impens F, Nahori MA, et al. (2013) A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science 341: 1238858. doi: 10.1126/science.1238858
|
| [49] |
Arbibe L, Kim DW, Batsche E, et al. (2007) An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat Immunol 8: 47-56. doi: 10.1038/ni1423
|
| [50] |
Harouz H, Rachez C, Meijer BM, et al. (2014) Shigella flexneri targets the HP1gamma subcode through the phosphothreonine lyase OspF. EMBO J 33: 2606-2622. doi: 10.15252/embj.201489244
|
| [51] |
Li H, Xu H, Zhou Y, et al. (2007) The phosphothreonine lyase activity of a bacterial type III effector family. Science 315: 1000-1003. doi: 10.1126/science.1138960
|
| [52] | Foster SL, Hargreaves DC, Medzhitov R (2007) Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447: 972-978. |
| [53] |
El Gazzar M, Liu T, Yoza BK, et al. (2010) Dynamic and selective nucleosome repositioning during endotoxin tolerance. J Biol Chem 285: 1259-1271. doi: 10.1074/jbc.M109.067330
|
| [54] |
Shalova IN, Lim JY, Chittezhath M, et al. (2015) Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1alpha. Immunity 42: 484-498. doi: 10.1016/j.immuni.2015.02.001
|
| [55] |
Cheng SC, Scicluna BP, Arts RJ, et al. (2016) Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol 17: 406-413. doi: 10.1038/ni.3398
|
| [56] |
Chen J, Ivashkiv LB (2010) IFN-gamma abrogates endotoxin tolerance by facilitating Toll-like receptor-induced chromatin remodeling. Proc Natl Acad Sci U S A 107: 19438-19443. doi: 10.1073/pnas.1007816107
|
| [57] | Tribouley J, Tribouley-Duret J, Appriou M (1978) [Effect of Bacillus Callmette Guerin (BCG) on the receptivity of nude mice to Schistosoma mansoni]. C R Seances Soc Biol Fil 172: 902-904. |
| [58] |
Kleinnijenhuis J, Quintin J, Preijers F, et al. (2014) Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun 6: 152-158. doi: 10.1159/000355628
|
| [59] |
van 't Wout JW, Poell R, van Furth R (1992) The role of BCG/PPD-activated macrophages in resistance against systemic candidiasis in mice. Scand J Immunol 36: 713-719. doi: 10.1111/j.1365-3083.1992.tb03132.x
|
| [60] |
Kleinnijenhuis J, Quintin J, Preijers F, et al. (2012) Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A 109: 17537-17542. doi: 10.1073/pnas.1202870109
|
| [61] |
Quintin J, Saeed S, Martens JH, et al. (2012) Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12: 223-232. doi: 10.1016/j.chom.2012.06.006
|
| [62] |
Ostuni R, Piccolo V, Barozzi I, et al. (2013) Latent enhancers activated by stimulation in differentiated cells. Cell 152: 157-171. doi: 10.1016/j.cell.2012.12.018
|
| [63] |
Bezman NA, Kim CC, Sun JC, et al. (2012) Molecular definition of the identity and activation of natural killer cells. Nat Immunol 13: 1000-1009. doi: 10.1038/ni.2395
|
| [64] |
Schlums H, Cichocki F, Tesi B, et al. (2015) Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 42: 443-456. doi: 10.1016/j.immuni.2015.02.008
|
| [65] |
Kagi D, Ledermann B, Burki K, et al. (1994) Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369: 31-37. doi: 10.1038/369031a0
|
| [66] |
Ferlazzo G, Tsang ML, Moretta L, et al. (2002) Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J Exp Med 195: 343-351. doi: 10.1084/jem.20011149
|
| [67] |
Xu HC, Grusdat M, Pandyra AA, et al. (2014) Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 40: 949-960. doi: 10.1016/j.immuni.2014.05.004
|
| [68] |
Bouchon A, Cella M, Grierson HL, et al. (2001) Activation of NK cell-mediated cytotoxicity by a SAP-independent receptor of the CD2 family. J Immunol 167: 5517-5521. doi: 10.4049/jimmunol.167.10.5517
|
| [69] |
Kruse PH, Matta J, Ugolini S, et al. (2014) Natural cytotoxicity receptors and their ligands. Immunol Cell Biol 92: 221-229. doi: 10.1038/icb.2013.98
|
| [70] |
Uhrberg M, Valiante NM, Shum BP, et al. (1997) Human diversity in killer cell inhibitory receptor genes. Immunity 7: 753-763. doi: 10.1016/S1074-7613(00)80394-5
|
| [71] |
O'Leary JG, Goodarzi M, Drayton DL, et al. (2006) T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol 7: 507-516. doi: 10.1038/ni1332
|
| [72] |
Sun JC, Beilke JN, Lanier LL (2009) Adaptive immune features of natural killer cells. Nature 457: 557-561. doi: 10.1038/nature07665
|
| [73] |
Min-Oo G, Lanier LL (2014) Cytomegalovirus generates long-lived antigen-specific NK cells with diminished bystander activation to heterologous infection. J Exp Med 211: 2669-2680. doi: 10.1084/jem.20141172
|
| [74] |
Lee J, Zhang T, Hwang I, et al. (2015) Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 42: 431-442. doi: 10.1016/j.immuni.2015.02.013
|
| [75] |
Calore F, Lovat F, Garofalo M (2013) Non-coding RNAs and cancer. Int J Mol Sci 14: 17085-17110. doi: 10.3390/ijms140817085
|
| [76] |
Nagano T, Fraser P (2011) No-nonsense functions for long noncoding RNAs. Cell 145: 178-181. doi: 10.1016/j.cell.2011.03.014
|
| [77] | Da Sacco L, Baldassarre A, Masotti A (2012) Bioinformatics tools and novel challenges in long non-coding RNAs (lncRNAs) functional analysis. Int J Mol Sci 13: 97-114. |
| [78] |
Wilusz JE, Sunwoo H, Spector DL (2009) Long noncoding RNAs: functional surprises from the RNA world. Genes Dev 23: 1494-1504. doi: 10.1101/gad.1800909
|
| [79] |
Kaikkonen MU, Lam MT, Glass CK (2011) Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc Res 90: 430-440. doi: 10.1093/cvr/cvr097
|
| [80] |
Wang KC, Chang HY (2011) Molecular mechanisms of long noncoding RNAs. Mol Cell 43: 904-914. doi: 10.1016/j.molcel.2011.08.018
|
| [81] |
Ng KW, Anderson C, Marshall EA, et al. (2016) Piwi-interacting RNAs in cancer: emerging functions and clinical utility. Mol Cancer 15: 5. doi: 10.1186/s12943-016-0491-9
|
| [82] |
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281-297. doi: 10.1016/S0092-8674(04)00045-5
|
| [83] |
Doench JG, Sharp PA (2004) Specificity of microRNA target selection in translational repression. Genes Dev 18: 504-511. doi: 10.1101/gad.1184404
|
| [84] |
Grimson A, Farh KK, Johnston WK, et al. (2007) MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91-105. doi: 10.1016/j.molcel.2007.06.017
|
| [85] | Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10: 126-139. |
| [86] | Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843-854. |
| [87] |
Wightman B, Ha I, Ruvkun G (1993) Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75: 855-862. doi: 10.1016/0092-8674(93)90530-4
|
| [88] |
Paladini L, Fabris L, Bottai G, et al. (2016) Targeting microRNAs as key modulators of tumor immune response. J Exp Clin Cancer Res 35: 103. doi: 10.1186/s13046-016-0375-2
|
| [89] |
Taganov KD, Boldin MP, Chang KJ, et al. (2006) NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 103: 12481-12486. doi: 10.1073/pnas.0605298103
|
| [90] |
O'Neill LA, Sheedy FJ, McCoy CE (2011) MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol 11: 163-175. doi: 10.1038/nri2957
|
| [91] |
Pathak S, Grillo AR, Scarpa M, et al. (2015) MiR-155 modulates the inflammatory phenotype of intestinal myofibroblasts by targeting SOCS1 in ulcerative colitis. Exp Mol Med 47: e164. doi: 10.1038/emm.2015.21
|
| [92] |
Bazzoni F, Rossato M, Fabbri M, et al. (2009) Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci U S A 106: 5282-5287. doi: 10.1073/pnas.0810909106
|
| [93] |
Androulidaki A, Iliopoulos D, Arranz A, et al. (2009) The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity 31: 220-231. doi: 10.1016/j.immuni.2009.06.024
|
| [94] |
Curtale G, Mirolo M, Renzi TA, et al. (2013) Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc Natl Acad Sci U S A 110: 11499-11504. doi: 10.1073/pnas.1219852110
|
| [95] |
McCoy CE, Sheedy FJ, Qualls JE, et al. (2010) IL-10 inhibits miR-155 induction by toll-like receptors. J Biol Chem 285: 20492-20498. doi: 10.1074/jbc.M110.102111
|
| [96] |
Rossato M, Curtale G, Tamassia N, et al. (2012) IL-10-induced microRNA-187 negatively regulates TNF-alpha, IL-6, and IL-12p40 production in TLR4-stimulated monocytes. Proc Natl Acad Sci U S A 109: E3101-3110. doi: 10.1073/pnas.1209100109
|
| [97] |
Sheedy FJ, Palsson-McDermott E, Hennessy EJ, et al. (2010) Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol 11: 141-147. doi: 10.1038/ni.1828
|
| [98] |
El Gazzar M, McCall CE (2010) MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J Biol Chem 285: 20940-20951. doi: 10.1074/jbc.M110.115063
|
| [99] |
El Gazzar M, Church A, Liu T, et al. (2011) MicroRNA-146a regulates both transcription silencing and translation disruption of TNF-alpha during TLR4-induced gene reprogramming. J Leukoc Biol 90: 509-519. doi: 10.1189/jlb.0211074
|
| [100] |
Tili E, Michaille JJ, Cimino A, et al. (2007) Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol 179: 5082-5089. doi: 10.4049/jimmunol.179.8.5082
|
| [101] | Renzi TA, Rubino M, Gornati L, et al. (2015) MiR-146b Mediates Endotoxin Tolerance in Human Phagocytes. Mediators Inflamm 2015: 145305. |
| [102] |
Magistri M, Faghihi MA, St Laurent G, 3rd, et al. (2012) Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet 28: 389-396. doi: 10.1016/j.tig.2012.03.013
|
| [103] |
Louro R, El-Jundi T, Nakaya HI, et al. (2008) Conserved tissue expression signatures of intronic noncoding RNAs transcribed from human and mouse loci. Genomics 92: 18-25. doi: 10.1016/j.ygeno.2008.03.013
|
| [104] |
Pandey RR, Mondal T, Mohammad F, et al. (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32: 232-246. doi: 10.1016/j.molcel.2008.08.022
|
| [105] | Wang X, Song X, Glass CK, et al. (2011) The long arm of long noncoding RNAs: roles as sensors regulating gene transcriptional programs. Cold Spring Harb Perspect Biol 3: a003756. |
| [106] |
Guttman M, Rinn JL (2012) Modular regulatory principles of large non-coding RNAs. Nature 482: 339-346. doi: 10.1038/nature10887
|
| [107] | Kretz M, Siprashvili Z, Chu C, et al. (2013) Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 493: 231-235. |
| [108] |
Gong C, Maquat LE (2011) lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature 470: 284-288. doi: 10.1038/nature09701
|
| [109] |
Carpenter S, Aiello D, Atianand MK, et al. (2013) A long noncoding RNA mediates both activation and repression of immune response genes. Science 341: 789-792. doi: 10.1126/science.1240925
|
| [110] | Rapicavoli NA, Qu K, Zhang J, et al. (2013) A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. Elife 2: e00762. |
| [111] |
Li Z, Chao TC, Chang KY, et al. (2014) The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proc Natl Acad Sci U S A 111: 1002-1007. doi: 10.1073/pnas.1313768111
|
| [112] | Krawczyk M, Emerson BM (2014) p50-associated COX-2 extragenic RNA (PACER) activates COX-2 gene expression by occluding repressive NF-kappaB complexes. Elife 3: e01776. |
| [113] |
Liu B, Sun L, Liu Q, et al. (2015) A cytoplasmic NF-kappaB interacting long noncoding RNA blocks IkappaB phosphorylation and suppresses breast cancer metastasis. Cancer Cell 27: 370-381. doi: 10.1016/j.ccell.2015.02.004
|
| [114] |
Murphy MB, Medvedev AE (2016) Long noncoding RNAs as regulators of Toll-like receptor signaling and innate immunity. J Leukoc Biol 99: 839-850. doi: 10.1189/jlb.2RU1215-575R
|
| [115] |
Li W, Notani D, Rosenfeld MG (2016) Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet 17: 207-223. doi: 10.1038/nrg.2016.4
|
| [116] |
Hah N, Murakami S, Nagari A, et al. (2013) Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 23: 1210-1223. doi: 10.1101/gr.152306.112
|
| [117] |
Melgar MF, Collins FS, Sethupathy P (2011) Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol 12: R113. doi: 10.1186/gb-2011-12-11-r113
|
| [118] |
Zhu Y, Sun L, Chen Z, et al. (2013) Predicting enhancer transcription and activity from chromatin modifications. Nucleic Acids Res 41: 10032-10043. doi: 10.1093/nar/gkt826
|
| [119] |
Arner E, Daub CO, Vitting-Seerup K, et al. (2015) Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347: 1010-1014. doi: 10.1126/science.1259418
|
| [120] |
Hah N, Benner C, Chong LW, et al. (2015) Inflammation-sensitive super enhancers form domains of coordinately regulated enhancer RNAs. Proc Natl Acad Sci U S A 112: E297-302. doi: 10.1073/pnas.1424028112
|
| [121] |
Kim TK, Hemberg M, Gray JM, et al. (2010) Widespread transcription at neuronal activity-regulated enhancers. Nature 465: 182-187. doi: 10.1038/nature09033
|
| [122] |
Alexopoulou L, Holt AC, Medzhitov R, et al. (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413: 732-738. doi: 10.1038/35099560
|
| [123] |
Lee J, Sayed N, Hunter A, et al. (2012) Activation of innate immunity is required for efficient nuclear reprogramming. Cell 151: 547-558. doi: 10.1016/j.cell.2012.09.034
|
| [124] |
Takahashi K, Tanabe K, Ohnuki M, et al. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861-872. doi: 10.1016/j.cell.2007.11.019
|
| [125] |
Takahashi K, Okita K, Nakagawa M, et al. (2007) Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc 2: 3081-3089. doi: 10.1038/nprot.2007.418
|
| [126] | Meng S, Zhou G, Gu Q, et al. (2016) Transdifferentiation Requires iNOS Activation: Role of RING1A S-Nitrosylation. Circ Res 119: e129-e138. |
| [127] |
Buenrostro JD, Giresi PG, Zaba LC, et al. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213-1218. doi: 10.1038/nmeth.2688
|
| [128] |
Mercer TR, Edwards SL, Clark MB, et al. (2013) DNase I-hypersensitive exons colocalize with promoters and distal regulatory elements. Nat Genet 45: 852-859. doi: 10.1038/ng.2677
|
| [129] |
Nagano T, Lubling Y, Yaffe E, et al. (2015) Single-cell Hi-C for genome-wide detection of chromatin interactions that occur simultaneously in a single cell. Nat Protoc 10: 1986-2003. doi: 10.1038/nprot.2015.127
|
| [130] |
Miyanari Y, Torres-Padilla ME (2012) Control of ground-state pluripotency by allelic regulation of Nanog. Nature 483: 470-473. doi: 10.1038/nature10807
|
| [131] |
Soucie EL, Weng Z, Geirsdottir L, et al. (2016) Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science 351: aad5510. doi: 10.1126/science.aad5510
|
| [132] |
Paul F, Arkin Y, Giladi A, et al. (2015) Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 163: 1663-1677. doi: 10.1016/j.cell.2015.11.013
|
| [133] |
Abraham BJ, Cui K, Tang Q, et al. (2013) Dynamic regulation of epigenomic landscapes during hematopoiesis. BMC Genomics 14: 193. doi: 10.1186/1471-2164-14-193
|
| [134] |
Olsson A, Venkatasubramanian M, Chaudhri VK, et al. (2016) Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 537: 698-702. doi: 10.1038/nature19348
|
| [135] |
Epelman S, Lavine KJ, Beaudin AE, et al. (2014) Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40: 91-104. doi: 10.1016/j.immuni.2013.11.019
|
| [136] | Italiani P, Boraschi D (2014) From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front Immunol 5: 514. |
| [137] |
Ganan-Gomez I, Wei Y, Starczynowski DT, et al. (2015) Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia 29: 1458-1469. doi: 10.1038/leu.2015.69
|
| [138] |
Lin CY, Loven J, Rahl PB, et al. (2012) Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151: 56-67. doi: 10.1016/j.cell.2012.08.026
|
| [139] |
Liu G, Gramling S, Munoz D, et al. (2011) Two novel BRM insertion promoter sequence variants are associated with loss of BRM expression and lung cancer risk. Oncogene 30: 3295-3304. doi: 10.1038/onc.2011.81
|
| [140] |
Kawauchi S, Calof AL, Santos R, et al. (2009) Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/-) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet 5: e1000650. doi: 10.1371/journal.pgen.1000650
|
| [141] |
Ballman KV (2015) Biomarker: Predictive or Prognostic? J Clin Oncol 33: 3968-3971. doi: 10.1200/JCO.2015.63.3651
|
| [142] |
Mehta S, Shelling A, Muthukaruppan A, et al. (2010) Predictive and prognostic molecular markers for cancer medicine. Ther Adv Med Oncol 2: 125-148. doi: 10.1177/1758834009360519
|
| [143] |
van Leeuwen MA, Westra J, Limburg PC, et al. (1995) Clinical significance of interleukin-6 measurement in early rheumatoid arthritis: relation with laboratory and clinical variables and radiological progression in a three year prospective study. Ann Rheum Dis 54: 674-677. doi: 10.1136/ard.54.8.674
|
| [144] | Knudsen LS, Klarlund M, Skjodt H, et al. (2008) Biomarkers of inflammation in patients with unclassified polyarthritis and early rheumatoid arthritis. Relationship to disease activity and radiographic outcome. J Rheumatol 35: 1277-1287. |
| [145] |
Klein-Wieringa IR, van der Linden MP, Knevel R, et al. (2011) Baseline serum adipokine levels predict radiographic progression in early rheumatoid arthritis. Arthritis Rheum 63: 2567-2574. doi: 10.1002/art.30449
|
| [146] | Lard LR, Roep BO, Toes RE, et al. (2004) Enhanced concentrations of interleukin 16 are associated with joint destruction in patients with rheumatoid arthritis. J Rheumatol 31: 35-39. |
| [147] |
Syversen SW, Goll GL, Haavardsholm EA, et al. (2008) A high serum level of eotaxin (CCL 11) is associated with less radiographic progression in early rheumatoid arthritis patients. Arthritis Res Ther 10: R28. doi: 10.1186/ar2381
|
| [148] |
Irizarry RA, Ladd-Acosta C, Wen B, et al. (2009) The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41: 178-186. doi: 10.1038/ng.298
|
| [149] |
Laird PW (2003) The power and the promise of DNA methylation markers. Nat Rev Cancer 3: 253-266. doi: 10.1038/nrc1045
|
| [150] | Luczak MW, Jagodzinski PP (2006) The role of DNA methylation in cancer development. Folia Histochem Cytobiol 44: 143-154. |
| [151] | Lu H, Liu X, Deng Y, et al. (2013) DNA methylation, a hand behind neurodegenerative diseases. Front Aging Neurosci 5: 85. |
| [152] |
Richardson B, Scheinbart L, Strahler J, et al. (1990) Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum 33: 1665-1673. doi: 10.1002/art.1780331109
|
| [153] |
Liu Y, Aryee MJ, Padyukov L, et al. (2013) Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 31: 142-147. doi: 10.1038/nbt.2487
|
| [154] |
Lin SY, Hsieh SC, Lin YC, et al. (2012) A whole genome methylation analysis of systemic lupus erythematosus: hypomethylation of the IL10 and IL1R2 promoters is associated with disease activity. Genes Immun 13: 214-220. doi: 10.1038/gene.2011.74
|
| [155] |
Yeung KS, Chung BH, Choufani S, et al. (2017) Genome-Wide DNA Methylation Analysis of Chinese Patients with Systemic Lupus Erythematosus Identified Hypomethylation in Genes Related to the Type I Interferon Pathway. PLoS One 12: e0169553. doi: 10.1371/journal.pone.0169553
|
| [156] |
Hashimoto Y, Zumwalt TJ, Goel A (2016) DNA methylation patterns as noninvasive biomarkers and targets of epigenetic therapies in colorectal cancer. Epigenomics 8: 685-703. doi: 10.2217/epi-2015-0013
|
| [157] |
Uhl B, Gevensleben H, Tolkach Y, et al. (2017) PITX2 DNA Methylation as Biomarker for Individualized Risk Assessment of Prostate Cancer in Core Biopsies. J Mol Diagn 19: 107-114. doi: 10.1016/j.jmoldx.2016.08.008
|
| [158] |
Lofton-Day C, Model F, Devos T, et al. (2008) DNA methylation biomarkers for blood-based colorectal cancer screening. Clin Chem 54: 414-423. doi: 10.1373/clinchem.2007.095992
|
| [159] |
Yang M, Park JY (2012) DNA methylation in promoter region as biomarkers in prostate cancer. Methods Mol Biol 863: 67-109. doi: 10.1007/978-1-61779-612-8_5
|
| [160] |
Chung W, Kwabi-Addo B, Ittmann M, et al. (2008) Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PLoS One 3: e2079. doi: 10.1371/journal.pone.0002079
|
| [161] |
Jiao Y, Shi C, Edil BH, et al. (2011) DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331: 1199-1203. doi: 10.1126/science.1200609
|
| [162] |
Dalgliesh GL, Furge K, Greenman C, et al. (2010) Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463: 360-363. doi: 10.1038/nature08672
|
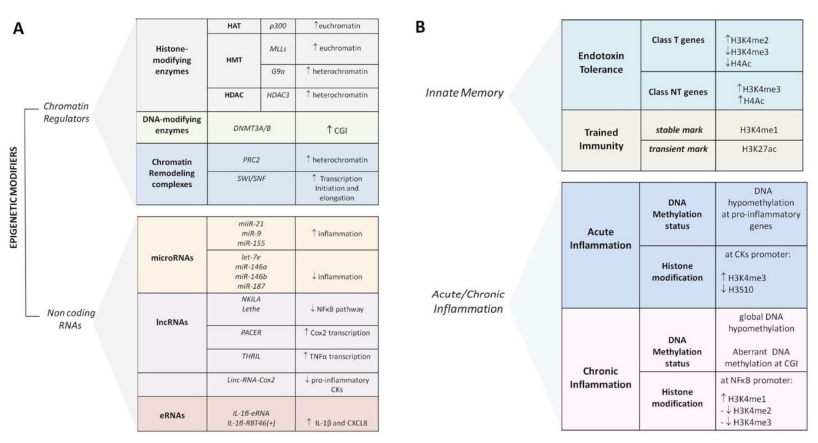
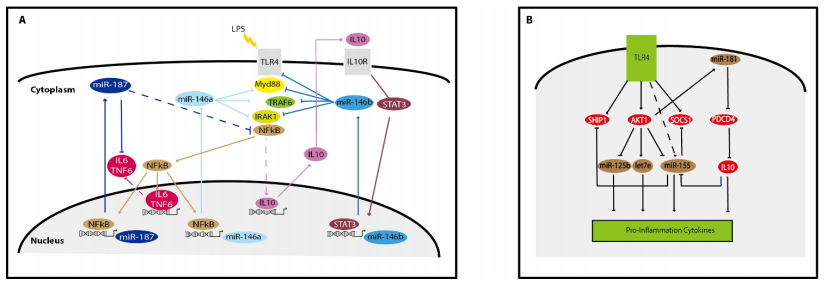
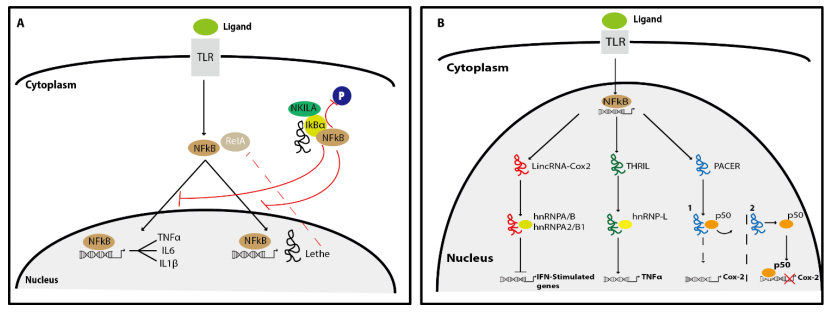
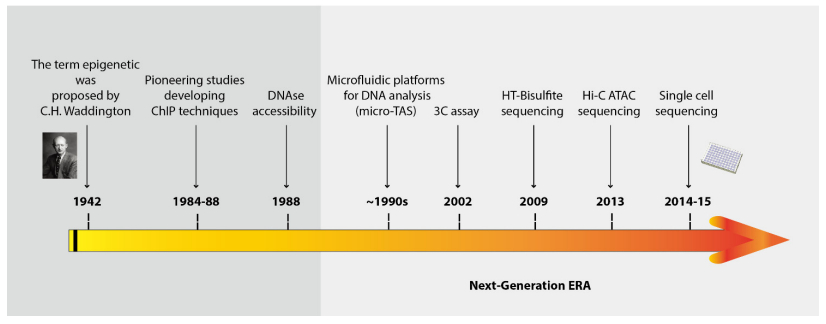
Figures(4) / Tables(2)

Mariacristina De Luca, Kevin Pels, Susana Moleirinho, Graziella Curtale. The epigenetic landscape of innate immunity[J]. AIMS Molecular Science, 2017, 4(1): 110-139. doi: 10.3934/molsci.2017.1.110







 DownLoad:
DownLoad: