Citation: Virginia L. Ma, Shili Lin. Examining the rare disease assumption used to justify HWE testing with control samples[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 73-91. doi: 10.3934/mbe.2020004

| [1] | J. Wittke-Thompson, A. Pluzhnikov and N. Cox, Rational inferences about departures from HardyWeinberg equilibrium, Am. J. Hum. Genet., 76 (2005), 967-986. |
| [2] | C. Yu, S. Zhang, C. Zhou, et al., A Likelihood Ratio Test of Population Hardy-Weinberg Equilibrium for Case-Control Studies, Genet. Epidemiol., 33 (2009), 275-280. |
| [3] | J. Wang and S. Shete, Testing Departure from Hardy-Weinberg Proportions, in Statistical Human Genetics: Methods and Protocols, 2nd Edition (ed. Elston, RC), vol. 1666 of Methods in Molecular Biology, Humana Press, 2017, 83-115. |
| [4] | I. Gomes, A. Collins, C. Lonjou, et al., Hardy-Weinberg quality control, Ann. Hum. Genet., 63 (1999), 535-538. |
| [5] | S. Weiss, E. Silverman and L. Palmer, Case-control association studies in pharmacogenetics, Pharmacogenomics J., 1 (2001), 157-158. |
| [6] | J. Xu, A. Turner, J. Little, et al., Positive results in association studies are associated with departure from Hardy-Weinberg equilibrium: hint for genotyping error? Hum. Genet., 111 (2002), 573-574. |
| [7] | L. Hosking, S. Lumsden, K. Lewis, et al., Detection of genotyping errors by Hardy-Weinberg equilibrium testing, Eur. J. Hum. Genet., 12 (2004), 395-399. |
| [8] | G. Salanti, G. Amountza, E. Ntzani, et al., Hardy-Weinberg equilibrium in genetic association studies: an empirical evaluation of reporting, deviations, and power, Eur. J. Hum. Genet., 13 (2005), 840-848. |
| [9] | R. Moonesinghe, A. Yesupriya, M.-h. Chang, et al., A Hardy-Weinberg Equilibrium Test for Analyzing Population Genetic Surveys With Complex Sample Designs, Am. J. Epidemiol., 171 (2010), 932-941. |
| [10] | S. Leal, Detection of genotyping errors and pseudo-SNPs via deviations from Hardy-Weinberg equilibrium, Genet. Epidemiol., 29 (2005), 204-214. |
| [11] | D. Cox and P. Kraft, Quantification of the power of Hardy-Weinberg equilibrium testing to detect genotyping error, Hum. Hered., 61 (2006), 10-14. |
| [12] | Y. Y. Teo, A. E. Fry, T. G. Clark, et al., On the Usage of HWE for Identifying Genotyping Errors, Ann. Hum. Genet., 71 (2007), 701-703. |
| [13] | G. Y. Zou and A. Donner, The merits of testing Hardy-Weinberg equilibrium in the analysis of unmatched case-control data: A cautionary note, Ann. Hum. Genet., 70 (2006), 923-933. |
| [14] | M. I. McCarthy, G. R. Abecasis, L. R. Cardon, et al., Genome-wide association studies for complex traits: consensus, uncertainty and challenges, Nat. Rev. Genet., 9 (2008), 356-369. |
| [15] | C. Healey, A. Dunning, M. Teare, et al., A common variant in BRCA2 is associated with both breast cancer risk and prenatal viability, Nat. Genet., 26 (2000), 362-364. |
| [16] | P. R. Burton, D. G. Clayton, L. R. Cardon, et al., Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls, Nature, 447 (2007), 661-678. |
| [17] | J. A. Phillips III, J. S. Poling, C. A. Phillips, et al., Synergistic heterozygosity for TGF beta 1 SNPs and BMPR2 mutations modulates the age at diagnosis and penetrance of familial pulmonary arterial hypertension, Genet. Med., 10 (2008), 359-365. |
| [18] | J. Wang and S. Shete, Using Both Cases and Controls for Testing Hardy-Weinberg Proportions in a Genetic Association Study, Hum. Hered., 69 (2010), 212-218. |
| [19] | D. Nielsen, M. Ehm and B. Weir, Detecting marker-disease association by testing for HardyWeinberg disequilibrium at a marker locus, Am J Hum Genet., 63 (1998), 1531-1540. |
| [20] | J. Graffelman and B. S. Weir, Testing for Hardy-Weinberg equilibrium at biallelic genetic markers on the X chromosome, Heredity, 116 (2016), 558-568. |
| [21] | C. C. Reyes-Gibby, J. Wang, S.-C. J. Yeung, et al., Genome-wide association study identifies genes associated with neuropathy in patients with head and neck cancer, Sci. Rep., 8. |
| [22] | M. Li and C. Li, Assessing Departure from Hardy-Weinberg Equilibrium in the Presence of Disease Association, Genet. Epidemiol., 32 (2008), 589-599. |
| [23] | J. Wang and S. Shete, Testing Hardy-Weinberg Proportions in a Frequency-Matched Case-Control Genetic Association Study, PLoS One, 6. |
| [24] | N. Chatterjee, Y.-H. Chen, S. Luo, et al., Analysis of Case-Control Association Studies: SNPs, Imputation and Haplotypes, Stat. Sci., 24 (2009), 489-502. |
| [25] | J. Wang, R. Yu and S. Shete, X-Chromosome Genetic Association Test Accounting for XInactivation, Skewed X-Inactivation, and Escape from X-Inactivation, Genet. Epidemiol., 38 (2014), 483-493. |
| [26] | Y. Zhang and Y. Yuan, A Shrinkage Method for Testing the Hardy-Weinberg Equilibrium in CaseControl Studies, Genet. Epidemiol., 37 (2013), 743-750. |
| [27] | M. Epstein and G. Satten, Inference on haplotype effects in case-control studies using unphased genotype data, Am. J. Hum. Genet., 73 (2003), 1316-1329. |
| [28] | D. G. Torgerson, E. J. Ampleford, G. Y. Chiu, et al., Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations, Nat. Genet., 43 (2011), 887-892. |
| [29] | P. K. Whelton, J. He and P. Muntner, Prevalence, awareness, treatment and control of hypertension in North America, North Africa and Asia, J. Hum. Hypertens., 18 (2004), 545-551. |
| [30] | S. Wild, G. Roglic, A. Green, et al., Global Prevalence of Diabetes, Am. Diabetes Assoc. Diabetes Care., 27 (2004), 1047-1053. |
| [31] | T. Richter, S. Nestler-Parr, R. Babela, et al., Rare Disease Terminology and Definitions-A Systematic Global Review: Report of the ISPOR Rare Disease Special Interest Group, Value in Health, 18 (2015), 906-914. |
| [32] | J. Yang and S. Lin, Robust Partial Likelihood Approach for Detecting Imprinting and Maternal Effects Using Case-Control Families, Ann. Appl. Stat., 7 (2013), 249-268. |
| [33] | A. Ziegler, S. Ghosh, T. D. Dyer, et al., Introduction to genetic analysis workshop 17 summaries, Genet. Epidemiol., 35 (2011), S1-S4. |
| [34] | M. W. Brems, The Rare Disease Assumption: The Good, The Bad, and The Ugly, Master's thesis, The Ohio State University, 2015. |
| [35] |
L. A. Torre, R. L. Siegel, E. M. Ward, et al., Global Cancer Incidence and Mortality Rates and Trends-An Update, Cancer Epidemiol. Biomark. Prev., 25 (2016), 16-27. doi: 10.1158/1055-9965.EPI-15-0578

|
| [36] | National Cancer Institute, Cancer Statistics, Natl Cancer Inst., 2019. Available from: https://www.cancer.gov/about-cancer/understanding/statistics. |
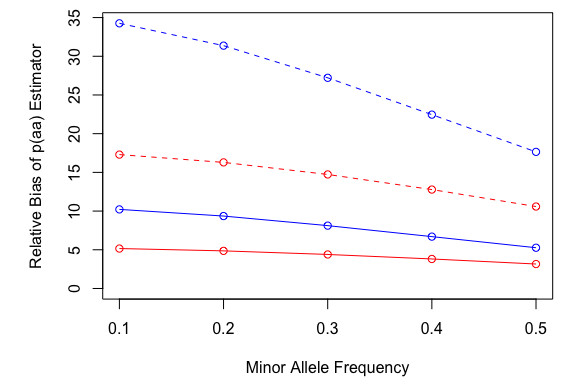
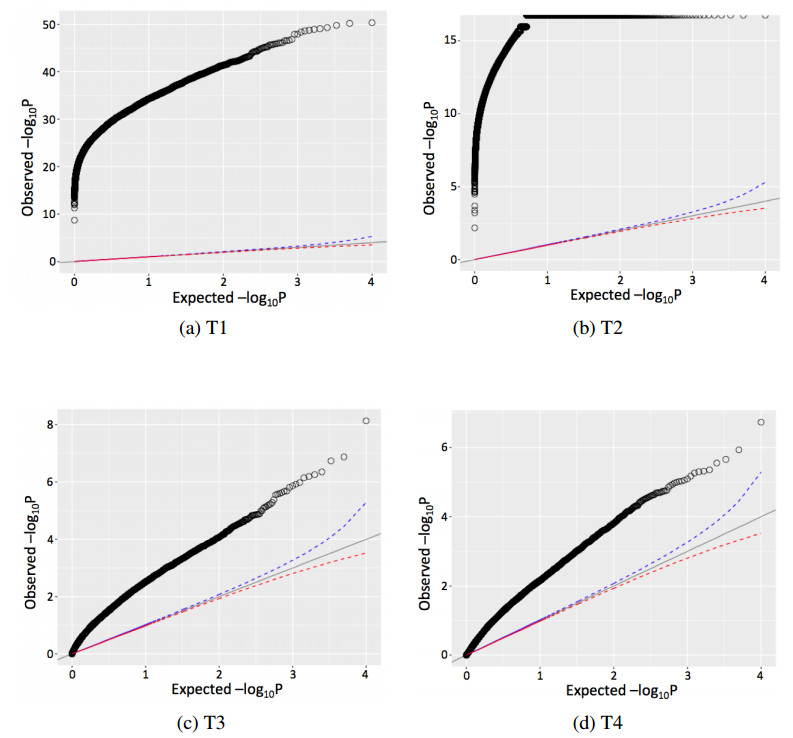
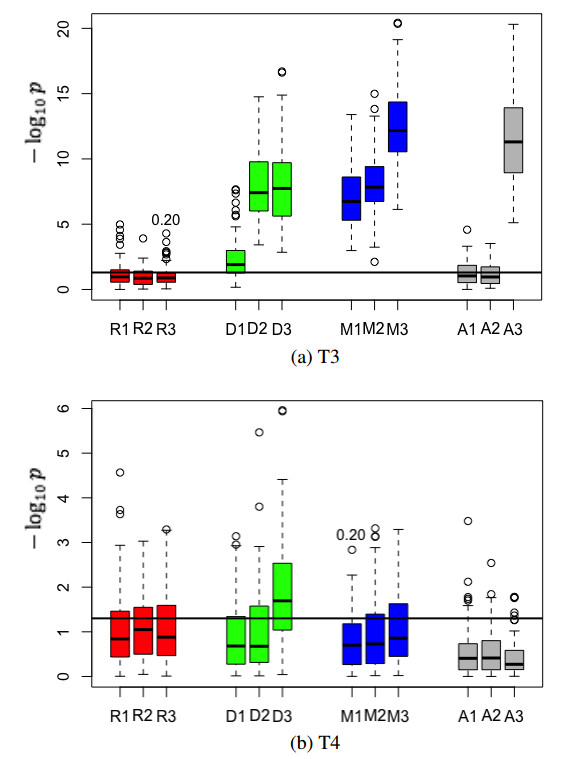
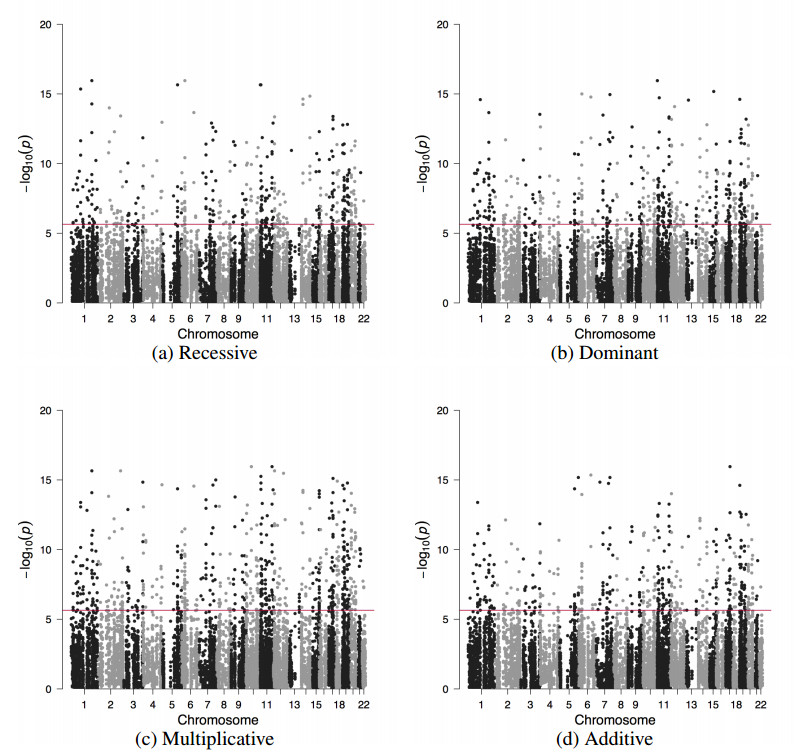
Figures(4) / Tables(2)

Virginia L. Ma, Shili Lin. Examining the rare disease assumption used to justify HWE testing with control samples[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 73-91. doi: 10.3934/mbe.2020004







 DownLoad:
DownLoad: