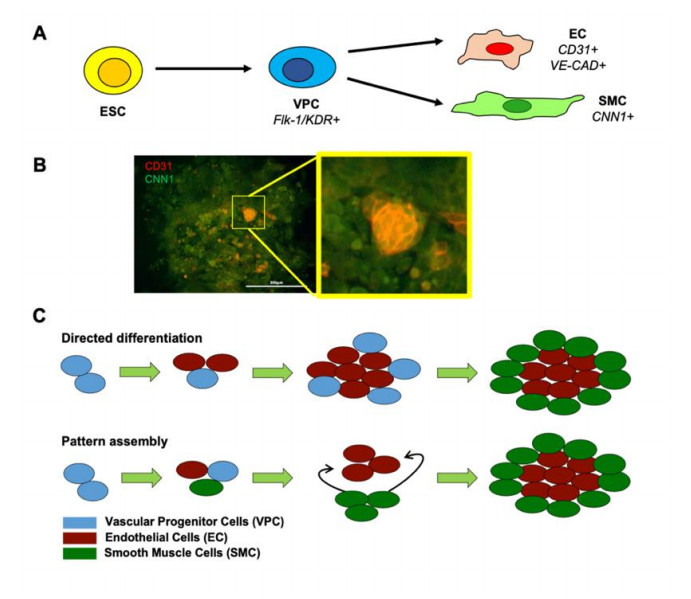
Vascular cells self-organize into unique structures guided by cell proliferation, migration, and/or differentiation from neighboring cells, mechanical factors, and/or soluble signals. However, the relative contribution of each of these factors remains unclear. Our objective was to develop a computational model to explore the different factors affecting the emerging micropatterns in 2D. This was accomplished by developing a stochastic on-lattice population-based model starting with vascular progenitor cells with the potential to proliferate, migrate, and/or differentiate into either endothelial cells or smooth muscle cells. The simulation results yielded patterns that were qualitatively and quantitatively consistent with experimental observations. Our results suggested that post-differentiation cell migration and proliferation when balanced could generate between 30–70% of each cell type enabling the formation of vascular patterns. Moreover, the cell-to-cell sensing could enhance the robustness of this patterning. These findings computationally supported that 2D patterning is mechanistically similar to current microfluidic platforms that take advantage of the migration-directed self-assembly of mature endothelial and mural cells to generate perfusable 3D vasculature in permissible hydrogel environments and suggest that stem or progenitor cells may not be fully necessary components in many tissue formations like those formed by vasculogenesis.
Citation: Jose E. Zamora Alvarado, Kara E. McCloskey, Ajay Gopinathan. Migration and proliferation drive the emergence of patterns in co-cultures of differentiating vascular progenitor cells[J]. Mathematical Biosciences and Engineering, 2024, 21(8): 6731-6757. doi: 10.3934/mbe.2024295
Vascular cells self-organize into unique structures guided by cell proliferation, migration, and/or differentiation from neighboring cells, mechanical factors, and/or soluble signals. However, the relative contribution of each of these factors remains unclear. Our objective was to develop a computational model to explore the different factors affecting the emerging micropatterns in 2D. This was accomplished by developing a stochastic on-lattice population-based model starting with vascular progenitor cells with the potential to proliferate, migrate, and/or differentiate into either endothelial cells or smooth muscle cells. The simulation results yielded patterns that were qualitatively and quantitatively consistent with experimental observations. Our results suggested that post-differentiation cell migration and proliferation when balanced could generate between 30–70% of each cell type enabling the formation of vascular patterns. Moreover, the cell-to-cell sensing could enhance the robustness of this patterning. These findings computationally supported that 2D patterning is mechanistically similar to current microfluidic platforms that take advantage of the migration-directed self-assembly of mature endothelial and mural cells to generate perfusable 3D vasculature in permissible hydrogel environments and suggest that stem or progenitor cells may not be fully necessary components in many tissue formations like those formed by vasculogenesis.

| [1] |
H. C. Ott, T. S. Matthiesen, S. K. Goh, L. D. Black, S. M. Kren, T. I. Netoff, et al., Perfusion-decellularized matrix: using nature's platform to engineer a bioartificial heart, Nat. Med., 14 (2008), 213–221. https://doi.org/10.1038/nm1684 doi: 10.1038/nm1684

|
| [2] |
J. M. Wainwright, C. A. Czajka, U. B. Patel, D. O. Freytes, K. Tobita, T. W. Gilbert, et al., Preparation of cardiac extracellular matrix from an intact porcine heart, Tissue Eng. Part C Methods, 16 (2010), 525–532. https://doi.org/10.1089/ten.tec.2009.0392 doi: 10.1089/ten.tec.2009.0392
|
| [3] |
A. B. Daly, J. M. Wallis, Z. D. Borg, R. W. Bonvillain, B. Deng, B. A. Ballif, et al., Initial binding and recellularization of decellularized mouse lung scaffolds with bone marrow-derived mesenchymal stromal cells, Tissue Eng. Part A, 18 (2012), 1–16. https://doi.org/10.1089/ten.tea.2011.0301 doi: 10.1089/ten.tea.2011.0301
|
| [4] |
J. Cortiella, J. Niles, A. Cantu, A. Brettler, A. Pham, G. Vargas, et al., Influence of acellular natural lung matrix on murine embryonic stem cell differentiation and tissue formation, Tissue Eng. Part A, 16 (2010), 2565–2580. https://doi.org/10.1089/ten.tea.2009.0730 doi: 10.1089/ten.tea.2009.0730
|
| [5] |
H. C. Ott, B. Clippinger, C. Conrad, C. Schuetz, I. Pomerantseva, L. Ikonomou, et al., Regeneration and orthotopic transplantation of a bioartificial lung, Nat. Med., 16 (2010), 927–933. https://doi.org/10.1038/nm.2193 doi: 10.1038/nm.2193
|
| [6] |
K. H. Nakayama, C. A. Batchelder, C. I. Lee, A. F. Tarantal, Decellularized rhesus monkey kidney as a three-dimensional scaffold for renal tissue engineering, Tissue Eng. Part A, 16 (2010), 2207–2216. https://doi.org/10.1089/ten.tea.2009.0602 doi: 10.1089/ten.tea.2009.0602
|
| [7] |
G. Orlando, C. Booth, Z. Wang, G. Totonelli, C. L. Ross, E. Moran, et al., Discarded human kidneys as a source of ECM scaffold for kidney regeneration technologies., Biomaterials, 34 (2013), 5915–25. https://doi.org/10.1016/j.biomaterials.2013.04.033 doi: 10.1016/j.biomaterials.2013.04.033
|
| [8] |
B. E. Uygun, A. Soto-Gutierrez, H. Yagi, M. L. Izamis, M. A. Guzzardi, C. Shulman, et al., Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix, Nat. Med., 16 (2010), 814–820. https://doi.org/10.1038/nm.2170 doi: 10.1038/nm.2170
|
| [9] |
A. Soto-Gutierrez, L. Zhang, C. Medberry, K. Fukumitsu, D. Faulk, H. Jiang, et al., A whole-organ regenerative medicine approach for liver replacement, Tissue Eng. Part C Methods, 17 (2011), 677–686. https://doi.org/10.1089/ten.tec.2010.0698 doi: 10.1089/ten.tec.2010.0698
|
| [10] |
Y. H. Hsu, M. L. Moya, C. C. W. Hughes, S. C. George, A. P. Lee, A microfluidic platform for generating large-scale nearly identical human microphysiological vascularized tissue arrays, Lab Chip, 13 (2013), 2990. https://doi.org/10.1039/c3lc50424g doi: 10.1039/c3lc50424g
|
| [11] |
K. Sakaguchi, T. Shimizu, S. Horaguchi, H. Sekine, M. Yamato, M. Umezu, et al., In vitro engineering of vascularized tissue surrogates, Sci. Rep., 3 (2013), 1316. https://doi.org/10.1038/srep01316 doi: 10.1038/srep01316
|
| [12] |
J. Zhang, L. Chu, Z. Hou, M. P. Schwartz, T. Hacker, V. Vickerman, et al., Functional characterization of human pluripotent stem cell-derived arterial endothelial cells, Proc. Natl. Acad. Sci., (2017), 201702295. https://doi.org/10.1073/pnas.1702295114 doi: 10.1073/pnas.1702295114
|
| [13] |
J. A. Whisler, M. B. Chen, R. D. Kamm, Control of perfusable microvascular network morphology using a multiculture microfluidic system, Tissue Eng. Part C Methods, 20 (2014), 543–552. https://doi.org/10.1089/ten.tec.2013.0370 doi: 10.1089/ten.tec.2013.0370
|
| [14] |
Z. Wan, S. Zhang, A. X. Zhong, S. E. Shelton, M. Campisi, S. K. Sundararaman, et al., A robust vasculogenic microfluidic model using human immortalized endothelial cells and Thy1 positive fibroblasts, Biomaterials, 276 (2021), 121032. https://doi.org/10.1016/j.biomaterials.2021.121032 doi: 10.1016/j.biomaterials.2021.121032
|
| [15] |
K. Yamamoto, K. Tanimura, M. Watanabe, H. Sano, H. Uwamori, Y. Mabuchi, et al., Construction of continuous capillary networks stabilized by pericyte-like perivascular cells, Tissue Eng. Part A, 25 (2019), 499–510. https://doi.org/10.1089/ten.tea.2018.0186 doi: 10.1089/ten.tea.2018.0186
|
| [16] |
S. Levenberg, J. S. Golub, M. Amit, J. Itskovitz-Eldor, R. Langer, Endothelial cells derived from human embryonic stem cells, Proc. Natl. Acad. Sci., 99 (2002), 4391–4396. https://doi.org/10.1073/pnas.032074999 doi: 10.1073/pnas.032074999
|
| [17] |
K. E. McCloskey, D. A. Smith, H. Jo, R. M. Nerem, Embryonic stem cell-derived endothelial cells may lack complete functional maturation in vitro, J. Vasc. Res., 43 (2006), 411–421. https://doi.org/10.1159/000094791 doi: 10.1159/000094791
|
| [18] |
Z. Gong, L. E. Niklason, Use of human mesenchymal stem cells as alternative source of smooth muscle cells in vessel engineering, Methods Mol. Biol., 698 (2011), 279–294. https://doi.org/10.1007/978-1-60761-999-4_21 doi: 10.1007/978-1-60761-999-4_21
|
| [19] |
G. K. Owens, Regulation of differentiation of vascular smooth muscle cells, Physiol. Rev., 75 (1995), 487–517. https://doi.org/10.1152/physrev.1995.75.3.487 doi: 10.1152/physrev.1995.75.3.487
|
| [20] |
E. M. Shen, K. E. McCloskey, Development of mural cells: From in vivo understanding to in vitro recapitulation, Stem Cells Dev., 26 (2017), 1020–1041. https://doi.org/10.1089/scd.2017.0020 doi: 10.1089/scd.2017.0020
|
| [21] |
B. Descamps, C. Emanueli, Vascular differentiation from embryonic stem cells: Novel technologies and therapeutic promises, Vascul. Pharmacol., 56 (2012), 267–279. https://doi.org/10.1016/j.vph.2012.03.007 doi: 10.1016/j.vph.2012.03.007
|
| [22] |
R. A. Wimmer, A. Leopoldi, M. Aichinger, N. Wick, B. Hantusch, M. Novatchkova, et al., Human blood vessel organoids as a model of diabetic vasculopathy, Nature, 565 (2019), 505–510. https://doi.org/10.1038/s41586-018-0858-8 doi: 10.1038/s41586-018-0858-8
|
| [23] |
A. A. Blancas, A. J. Shih, N. E. Lauer, K. E. McCloskey, Endothelial cells from embryonic stem cells in a chemically defined medium, Stem Cells Dev., 20 (2011), 2153–2161. https://doi.org/10.1089/scd.2010.0432 doi: 10.1089/scd.2010.0432
|
| [24] |
K. McCloskey, D. Glaser, A. Burns, R. Hatano, Y. Fan, M. Medrzycki, Specialized mouse embryonic stem cells for studying vascular development, Stem Cells Cloning Adv. Appl., 7 (2014), 79. https://doi.org/10.2147/SCCAA.S69554 doi: 10.2147/SCCAA.S69554
|
| [25] |
D. E. Glaser, W. S. Turner, N. Madfis, L. Wong, J. Zamora, N. White, et al., Multifactorial optimizations for directing endothelial fate from stem cells, PLoS One, 11 (2016), e0166663. https://doi.org/10.1371/journal.pone.0166663 doi: 10.1371/journal.pone.0166663
|
| [26] |
B. Jahan, K. E. McCloskey, Differentiation and expansion of endothelial cells requires pre-optimization of KDR+ expression kinetics, Stem Cell Res., 42 (2020), 101685. https://doi.org/10.1016/j.scr.2019.101685 doi: 10.1016/j.scr.2019.101685
|
| [27] |
L. Wong, A. Kumar, B. Gabela-Zuniga, J. Chua, G. Singh, C. L. Happe, et al., Substrate stiffness directs diverging vascular fates, Acta Biomater., 96 (2019), 321–329. https://doi.org/10.1016/j.actbio.2019.07.030 doi: 10.1016/j.actbio.2019.07.030
|
| [28] |
J. M. Osborne, A. G. Fletcher, J. M. Pitt-Francis, P. K. Maini, D. J. Gavaghan, Comparing individual-based approaches to modelling the self-organization of multicellular tissues, PLOS Comput. Biol., 13 (2017), e1005387. https://doi.org/10.1371/journal.pcbi.1005387 doi: 10.1371/journal.pcbi.1005387
|
| [29] |
D. Viens, A three-dimensional finite element model for the mechanics of cell-cell interactions, J. Biomech. Eng., 129 (2007), 651. https://doi.org/10.1115/1.2768375 doi: 10.1115/1.2768375
|
| [30] |
G. W. Brodland, D. A. Clausi, Embryonic tissue morphogenesis modeled by FEM, J. Biomech. Eng., 116 (1994), 146–155. https://doi.org/10.1115/1.2895713 doi: 10.1115/1.2895713
|
| [31] |
Q. Smith, E. Stukalin, S. Kusuma, S. Gerecht, S. X. Sun, Stochasticity and spatial interaction govern stem cell differentiation dynamics, Sci. Rep., 5 (2015), 1–10. https://doi.org/10.1038/srep12617 doi: 10.1038/srep12617
|
| [32] |
W. C. Lo, C. S. Chou, K. Gokoffski, F. Wan, A. Lander, A. Calof, et al., Feedback regulation in multistage cell lineages, Math. Biosci. Eng., 6 (2009), 59–82. https://doi.org/10.3934/mbe.2009.6.59 doi: 10.3934/mbe.2009.6.59
|
| [33] |
A. Szabó, R. Ünnep, E. Méhes, W. O. Twal, W. S. Argraves, Y. Cao, et al., Collective cell motion in endothelial monolayers, Phys. Biol., 7 (2010), 046007. https://doi.org/10.1088/1478-3975/7/4/046007 doi: 10.1088/1478-3975/7/4/046007
|
| [34] |
Y. Mao, A. L. Tournier, P. A. Bates, J. E. Gale, N. Tapon, B. J. Thompson, Planar polarization of the atypical myosin Dachs orients cell divisions in Drosophila, Genes Dev., 25 (2011), 131–136. https://doi.org/10.1101/gad.610511 doi: 10.1101/gad.610511
|
| [35] |
D. C. Walker, G. Hill, S. M. Wood, R. H. Smallwood, J. Southgate, Agent-based computational modeling of wounded epithelial cell monolayers, IEEE Trans. Nanobiosci., 3 (2004), 153–163. https://doi.org/10.1109/TNB.2004.833680 doi: 10.1109/TNB.2004.833680
|
| [36] |
E. L. Bearer, J. S. Lowengrub, H. B. Frieboes, Y. L. Chuang, F. Jin, S. M. Wise, et al., Multiparameter computational modeling of ttumor invasion, Cancer Res., 69 (2009), 4493–4501. https://doi.org/10.1158/0008-5472.CAN-08-3834 doi: 10.1158/0008-5472.CAN-08-3834
|
| [37] |
D. Drasdo, S. Höhme, A single-cell-based model of tumor growth in vitro : monolayers and spheroids, Phys. Biol., 2 (2005), 133–147. https://doi.org/10.1088/1478-3975/2/3/001 doi: 10.1088/1478-3975/2/3/001
|
| [38] |
C. S. Chou, W. C. Lo, K. K. Gokoffski, Y. T. Zhang, F. Y. M. Wan, A. D. Lander, et al., Spatial dynamics of multistage cell lineages in tissue stratification, Biophys. J., 99 (2010), 3145–3154. https://doi.org/10.1016/j.bpj.2010.09.034 doi: 10.1016/j.bpj.2010.09.034
|
| [39] |
D. J. Kelly, P. J. Prendergast, Mechano-regulation of stem cell differentiation and tissue regeneration in osteochondral defects, J. Biomech., 38 (2005), 1413–1422. https://doi.org/10.1016/j.jbiomech.2004.06.026 doi: 10.1016/j.jbiomech.2004.06.026
|
| [40] |
H. Du, Y. Wang, D. Haensel, B. Lee, X. Dai, Q. Nie, Multiscale modeling of layer formation in epidermis, PLOS Comput. Biol., 14 (2018), e1006006. https://doi.org/10.1371/journal.pcbi.1006006 doi: 10.1371/journal.pcbi.1006006
|
| [41] |
A. Atala, Re: Collective and single cell behavior in epithelial contact inhibition, J. Urol., 188 (2012), 1396–1397. https://doi.org/10.1016/j.juro.2012.06.073 doi: 10.1016/j.juro.2012.06.073
|
| [42] |
B. Schreier, G. Schwerdt, C. Heise, D. Bethmann, S. Rabe, S. Mildenberger, et al., Substance-specific importance of EGFR for vascular smooth muscle cells motility in primary culture, Biochim. Biophys. Acta Mol. Cell Res., 1863 (2016), 1519–1533. https://doi.org/10.1016/j.bbamcr.2016.03.017 doi: 10.1016/j.bbamcr.2016.03.017
|
| [43] |
J. Walter-Yohrling, S. Morgenbesser, C. Rouleau, R. Bagley, M. Callahan, W. Weber, et al., Murine endothelial cell lines as models of tumor endothelial cells, Clin. Cancer Res., 10 (2004), 2179–2189. https://doi.org/10.1158/1078-0432.CCR-03-1013 doi: 10.1158/1078-0432.CCR-03-1013
|
| [44] |
N. Endlich, K. Endlich, N. Taesch, J. J. Helwig, Culture of vascular smooth muscle cells from small arteries of the rat kidney, Kidney Int., 57 (2000), 2468–2475. https://doi.org/10.1046/j.1523-1755.2000.00105.x doi: 10.1046/j.1523-1755.2000.00105.x
|
| [45] |
S. I. Nishikawa, S. Nishikawa, M. Hirashima, N. Matsuyoshi, H. Kodama, Progressive lineage analysis by cell sorting and culture identifies FLK1+VE-cadherin+ cells at a diverging point of endothelial and hemopoietic lineages, Development, 125 (1998), 1747–1757. https://doi.org/10.1111/ijpo.259 doi: 10.1111/ijpo.259
|
| [46] |
J. Yamashita, H. Itoh, M. Hirashima, M. Ogawa, S. Nishikawa, T. Yurugi, et al., Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors, Nature, 408 (2000), 92–96. https://doi.org/10.1038/35040568 doi: 10.1038/35040568
|
| [47] |
K. L. Hill, P. Obrtlikova, D. F. Alvarez, J. A. King, S. A. Keirstead, J. R. Allred, et al., Human embryonic stem cell−derived vascular progenitor cells capable of endothelial and smooth muscle cell function, Exp. Hematol., 38 (2010), 246–257. https://doi.org/10.1016/j.exphem.2010.01.001 doi: 10.1016/j.exphem.2010.01.001
|
| [48] | G. Van Rossum, F. L. Drake Jr, Python reference manual, Department of Computer Science, CWI, 1995. |
| [49] | MATLAB, version 9.1.0.441655 (R2016b), Natick, MA: The MathWorks, Inc. Available from: https://www.mathworks.com |
| [50] |
A. Huttenlocher, M. Lakonishok, M. Kinder, S. Wu, T. Truong, K. A. Knudsen, et al., Integrin and cadherin synergy regulates contact inhibition of migration and motile activity, J. Cell Biol., 141 (1998), 515–526. https://doi.org/10.1083/jcb.141.2.515 doi: 10.1083/jcb.141.2.515
|
| [51] |
M. Abercrombie, J. E. M. Heaysman, Observations on the social behaviour of cells in tissue culture, Exp. Cell Res., 6 (1954), 293–306. https://doi.org/10.1016/0014-4827(54)90176-7 doi: 10.1016/0014-4827(54)90176-7
|
| [52] |
M. S. Steinberg, Does differential adhesion govern self‐assembly processes in histogenesis? Equilibrium configurations and the emergence of a hierarchy among populations of embryonic cells, J. Exp. Zool., 173 (1970), 395–433. https://doi.org/10.1002/jez.1401730406 doi: 10.1002/jez.1401730406
|
| [53] |
B. Lilly, We have contact: endothelial cell-smooth muscle cell interactions, Physiology, 29 (2014), 234–241. https://doi.org/10.1152/physiol.00047.2013 doi: 10.1152/physiol.00047.2013
|
| [54] |
W. Baumgartner, P. Hinterdorfer, W. Ness, A. Raab, D. Vestweber, H. Schindler, et al., Cadherin interaction probed by atomic force microscopy, Proc. Natl. Acad. Sci., 97 (2000), 4005–4010. https://doi.org/10.1073/pnas.070052697 doi: 10.1073/pnas.070052697
|
| [55] |
E. Moiseeva, Adhesion receptors of vascular smooth muscle cells and their functions, Cardiovasc. Res., 52 (2001), 372–386. https://doi.org/10.1016/S0008-6363(01)00399-6 doi: 10.1016/S0008-6363(01)00399-6
|
| [56] |
E. Perret, A. Leung, H. Feracci, E. Evans, Trans-bonded pairs of E-cadherin exhibit a remarkable hierarchy of mechanical strengths, Proc. Natl. Acad. Sci., 101 (2004), 16472–16477. https://doi.org/10.1073/pnas.0402085101 doi: 10.1073/pnas.0402085101
|
| [57] |
M. Noseda, L. Chang, G. McLean, J. E. Grim, B. E. Clurman, L. L. Smith, et al., Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: role of p21Cip1 repression, Mol. Cell. Biol., 24 (2004), 8813–8822. https://doi.org/10.1128/MCB.24.20.8813-8822.2004 doi: 10.1128/MCB.24.20.8813-8822.2004
|
| [58] |
J. P. Sasine, K. T. Yeo, J. P. Chute, Concise review: Paracrine functions of vascular niche cells in regulating hematopoietic stem cell fate, Stem Cells Transl. Med., 6 (2017), 482–489. https://doi.org/https://doi.org/10.5966/sctm.2016-0254 doi: 10.5966/sctm.2016-0254
|
| [59] |
H. L. Kirschenlohr, J. C. Metcalfe, P. L. Weissberg, D. J. Grainger, Adult human aortic smooth muscle cells in culture produce active TGF-beta, Am. J. Physiol., 265 (1993), C571–C576. https://doi.org/10.1152/ajpcell.1993.265.2.C571 doi: 10.1152/ajpcell.1993.265.2.C571
|
| [60] |
H. Huang, X. Zhao, L. Chen, C. Xu, X. Yao, Y. Lu, et al., Differentiation of human embryonic stem cells into smooth muscle cells in adherent monolayer culture, Biochem. Biophys. Res. Commun., 351 (2006), 321–327. https://doi.org/10.1016/j.bbrc.2006.09.171 doi: 10.1016/j.bbrc.2006.09.171
|
| [61] |
E. G. Rens, M. T. Zeegers, I. Rabbers, A. Szabó, R. M. H. Merks, Autocrine inhibition of cell motility can drive epithelial branching morphogenesis in the absence of growth, Philos. Trans. R. Soc. B Biol. Sci., 375 (2020), 20190386. https://doi.org/10.1098/rstb.2019.0386 doi: 10.1098/rstb.2019.0386
|
| [62] |
S. H. Yoon, Y. K. Kim, E. D. Han, Y. H. Seo, B. H. Kim, M. R. K. Mofrad, Passive control of cell locomotion using micropatterns: the effect of micropattern geometry on the migratory behavior of adherent cells, Lab Chip, 12 (2012), 2391. https://doi.org/10.1039/c2lc40084g doi: 10.1039/c2lc40084g
|
| [63] |
A. Spradling, D. Drummond-Barbosa, T. Kai, Stem cells find their niche, Nature, 414 (2001), 98–104. https://doi.org/10.1038/35102160 doi: 10.1038/35102160
|
| [64] |
P. J. Albert, U. S. Schwarz, Modeling cell shape and dynamics on micropatterns, Cell Adh. Migr., 10 (2016), 516–528. https://doi.org/10.1080/19336918.2016.1148864 doi: 10.1080/19336918.2016.1148864
|
| [65] |
X. Jiang, D. A. Bruzewicz, A. P. Wong, M. Piel, G. M. Whitesides, Directing cell migration with asymmetric micropatterns, Proc. Natl. Acad. Sci., 102 (2005), 975–978. https://doi.org/10.1073/pnas.0408954102 doi: 10.1073/pnas.0408954102
|
| [66] |
N. Ojeh, I. Pastar, M. Tomic-Canic, O. Stojadinovic, Stem cells in skin regeneration, wound healing, and their clinical applications, Int. J. Mol. Sci., 16 (2015), 25476–25501. https://doi.org/10.3390/ijms161025476 doi: 10.3390/ijms161025476
|
| [67] |
R. I. Johnson, Hexagonal patterning of the Drosophila eye, Dev. Biol., 478 (2021), 173–182. https://doi.org/10.1016/j.ydbio.2021.07.004 doi: 10.1016/j.ydbio.2021.07.004
|
 mbe-21-08-295-Supplementary.pdf mbe-21-08-295-Supplementary.pdf |
 |






Figures(7) / Tables(1)

Jose E. Zamora Alvarado, Kara E. McCloskey, Ajay Gopinathan. Migration and proliferation drive the emergence of patterns in co-cultures of differentiating vascular progenitor cells[J]. Mathematical Biosciences and Engineering, 2024, 21(8): 6731-6757. doi: 10.3934/mbe.2024295







 DownLoad:
DownLoad: