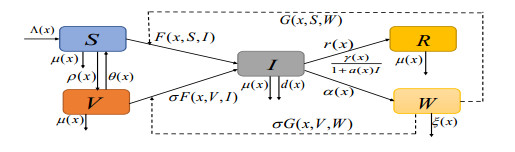
With the consideration of the complexity of the transmission of Cholera, a partially degenerated reaction-diffusion model with multiple transmission pathways, incorporating the spatial heterogeneity, general incidence, incomplete immunity, and Holling type Ⅱ treatment was proposed. First, the existence, boundedness, uniqueness, and global attractiveness of solutions for this model were investigated. Second, one obtained the threshold condition $ \mathcal{R}_{0} $ and gave its expression, which described global asymptotic stability of disease-free steady state when $ \mathcal{R}_{0} < 1 $, as well as the maximum treatment rate as zero. Further, we obtained the disease was uniformly persistent when $ \mathcal{R}_{0} > 1 $. Moreover, one used the mortality due to disease as a branching parameter for the steady state, and the results showed that the model undergoes a forward bifurcation at $ \mathcal{R}_{0} $ and completely excludes the presence of endemic steady state when $ \mathcal{R}_{0} < 1 $. Finally, the theoretical results were explained through examples of numerical simulations.
Citation: Shengfu Wang, Linfei Nie. Global analysis of a diffusive Cholera model with multiple transmission pathways, general incidence and incomplete immunity in a heterogeneous environment[J]. Mathematical Biosciences and Engineering, 2024, 21(4): 4927-4955. doi: 10.3934/mbe.2024218
With the consideration of the complexity of the transmission of Cholera, a partially degenerated reaction-diffusion model with multiple transmission pathways, incorporating the spatial heterogeneity, general incidence, incomplete immunity, and Holling type Ⅱ treatment was proposed. First, the existence, boundedness, uniqueness, and global attractiveness of solutions for this model were investigated. Second, one obtained the threshold condition $ \mathcal{R}_{0} $ and gave its expression, which described global asymptotic stability of disease-free steady state when $ \mathcal{R}_{0} < 1 $, as well as the maximum treatment rate as zero. Further, we obtained the disease was uniformly persistent when $ \mathcal{R}_{0} > 1 $. Moreover, one used the mortality due to disease as a branching parameter for the steady state, and the results showed that the model undergoes a forward bifurcation at $ \mathcal{R}_{0} $ and completely excludes the presence of endemic steady state when $ \mathcal{R}_{0} < 1 $. Finally, the theoretical results were explained through examples of numerical simulations.

| [1] |
R. Colwell, A. Huq, Environmental reservoir of Vibrio cholerae, the causative agent of cholera, Ann. Ny. Acad. Sci., 740 (1994), 44–53. https://doi.org/10.1111/j.1749-6632.1994.tb19852.x doi: 10.1111/j.1749-6632.1994.tb19852.x

|
| [2] |
A. A. Weil, A. I. Khan, F. Chowdhury, R. C. LaRocque, A. S. G. Faruque, E. T. Ryan, et al., Clinical outcomes of household contacts of patients with cholera in Bangladesh, Clin. Infect. Dis., 49 (2009), 1473–1479. https://doi.org/10.1086/644779 doi: 10.1086/644779
|
| [3] | World Health Organization, Cholera. Available from: https://www.who.int/news-room/fact-sheets/detail/cholera. |
| [4] | World Health Organization – EMRO – Yemen cholera situation reports. Available from: https://www.emro.who.int/yem/yemeninfocus/situation-reports.html. |
| [5] |
V. Rouzier, K. Severe, M. A. Juste, M. Peck, C. Perodin, P. Severe, et al., Cholera vaccination inurban Haiti, Am. J. Trop. Med. Hyg., 89 (2013), 671–681. https://doi.org/10.4269/ajtmh.13-0171 doi: 10.4269/ajtmh.13-0171
|
| [6] |
J. Andrews, S. Basu, Transmission dynamics and control of cholera in Haiti: an epidemic model, Lancet, 377 (2011), 1248–1255. https://doi.org/10.1016/S0140-6736(11)60273-0 doi: 10.1016/S0140-6736(11)60273-0
|
| [7] |
M. C. Eisenberg, Z. S. Shuai, J. H. Tien, P. van den Driessche, A cholera model in a patchy environment with water and human movement, Math. Biosci., 246 (2013), 105–112. https://doi.org/10.1016/j.mbs.2013.08.003 doi: 10.1016/j.mbs.2013.08.003
|
| [8] |
C. W. Song, R. Xu, A note on the global stability of a multi-strain cholera model with an imperfect vaccine, Appl. Math. Lett., 134 (2022), 108326. https://doi.org/10.1016/j.aml.2022.108326 doi: 10.1016/j.aml.2022.108326
|
| [9] |
X. Y. Zhou, X. Y. Shi, J. A. Cui, Stability and backward bifurcation on a cholera epidemic model with saturated recovery rate, Math. Method Appl. Sci., 40 (2017), 1288–306. https://doi.org/10.1002/mma.4053 doi: 10.1002/mma.4053
|
| [10] |
D. H. He, X. Y. Wang, D. Z. Gao, J. Wang, Modeling the 2016-2017 Yemen cholera outbreak with the impact of limited medical resources, J. Theor. Biol., 451 (2018), 80–85. https://doi.org/10.1016/j.jtbi.2018.04.041 doi: 10.1016/j.jtbi.2018.04.041
|
| [11] |
C. Y. Yang, J. Wang, On the intrinsic dynamics of bacteria in waterborne infections, Math. Biosci., 296 (2018), 71–81. https://doi.org/10.1016/j.mbs.2017.12.005 doi: 10.1016/j.mbs.2017.12.005
|
| [12] |
H. M. N. Teytsa, B. Tsanou, S. Bowong, J. Lubuma, Coupling the modeling of phage-bacteria interaction and cholera epidemiological model with and without optimal control, J. Theor. Biol., 512 (2021), 110537. https://doi.org/10.1016/j.jtbi.2020.110537 doi: 10.1016/j.jtbi.2020.110537
|
| [13] |
J. Z. Lin, X. Rui, X. H. Tian, Transmission dynamics of cholera with hyperinfectious and hypoinfectious vibrios: mathematical modelling and control strategies, Math. Biosci. Eng., 16 (2019), 4339–4358. http://dx.doi.org/10.3934/mbe.2019216 doi: 10.3934/mbe.2019216
|
| [14] |
J. Y. Yang, C. Modnak, J. Wang, Dynamical analysis and optimal control simulation for an age-structured cholera transmission model, J. Franklin I., 356 (2019), 8438–8467. https://doi.org/10.1016/j.jfranklin.2019.08.016 doi: 10.1016/j.jfranklin.2019.08.016
|
| [15] |
J. H. Tien, D. J. D. Earn, Multiple transmission pathways and disease dynamics in a waterborne pathogen model, B. Math. Biol., 72 (2010), 1506–1533. https://doi.org/10.1007/s11538-010-9507-6 doi: 10.1007/s11538-010-9507-6
|
| [16] |
Y. Shi, J.G. Gao, J.L. Wang, Analysis of a reaction-diffusion host-pathogen model with horizontal transmission, J. Math. Anal. Appl., 481 (2020), 123481. https://doi.org/10.1016/j.jmaa.2019.123481 doi: 10.1016/j.jmaa.2019.123481
|
| [17] |
F. Capone, V. De Cataldis, R. De Luca, Influence of diffusion on the stability of equilibria in a reaction-diffusion system modeling cholera dynamic, J. Math. Biol., 71 (2015), 1107–1131. https://doi.org/10.1007/s00285-014-0849-9 doi: 10.1007/s00285-014-0849-9
|
| [18] |
E. Avila-Vales, Á. G. C. Pérez, Dynamics of a reaction-diffusion SIRS model with general incidence rate in a heterogeneous environment, Z. Angew. Math. Phys., 73 (2022), 1–23. https://doi.org/10.1007/s00033-021-01645-0 doi: 10.1007/s00033-021-01645-0
|
| [19] |
F. B. Wang, J. P. Shi, X. F. Zou, Dynamics of a host-pathogen system on a bounded spatial domain, Commun. Pur. Appl. Anal., 14 (2015), 2535–2560. https://doi.org/10.3934/cpaa.2015.14.2535 doi: 10.3934/cpaa.2015.14.2535
|
| [20] |
E. Avila-Vales, G. E. Garcia-Almeida, Á. G. C. Pérez, Qualitative analysis of a diffusive SIR epidemic model with saturated incidence rate in a heterogeneous environment, J. Math. Anal. Appl., 503 (2021), 125295. https://doi.org/10.1016/j.jmaa.2021.125295 doi: 10.1016/j.jmaa.2021.125295
|
| [21] |
J. L. Wang, F. L. Xie, T. Kuniya, Analysis of a reaction-diffusion cholera epidemic model in a spatially heterogeneous environment, Commun. Nonlinear Sci., 80, (2020), 104951. https://doi.org/10.1016/j.cnsns.2019.104951 doi: 10.1016/j.cnsns.2019.104951
|
| [22] |
X. D. Chen, R. H. Cui, Global stability in a diffusive cholera epidemic model with nonlinear incidence, Appl. Math. Lett., 111 (2021), 106596. https://doi.org/10.1016/j.aml.2020.106596 doi: 10.1016/j.aml.2020.106596
|
| [23] |
Y. Yang, L. Zou, J. L. Zhou, C. H. Hsu, Dynamics of a waterborne pathogen model with spatial heterogeneity and general incidence rate, Nonlinear Anal.-Real., 53 (2020), 103065. https://doi.org/10.1016/j.nonrwa.2019.103065 doi: 10.1016/j.nonrwa.2019.103065
|
| [24] |
X. Y. Wang, F. B. Wang, Impact of bacterial hyperinfectivity on cholera epidemics in a spatially heterogeneous environment, J. Math. Anal. Appl., 480 (2019), 123407. https://doi.org/10.1016/j.jmaa.2019.123407 doi: 10.1016/j.jmaa.2019.123407
|
| [25] |
J. L. Wang, X. Q. Wu, Dynamics and profiles of a diffusive cholera model with bacterial hyperinfectivity and distinct dispersal rates, J. Dyn. Differ. Equations, 35 (2023), 1205–1241. http://dx.doi.org/10.1007/s10884-021-09975-3 doi: 10.1007/s10884-021-09975-3
|
| [26] |
D. M. Hartley, J. G. Morris Jr, D. L. Smith, Hyperinfectivity: a criticalelement in the ability of V.cholerae to cause epidemics?, Plos Med., 3 (2006), e7. https://doi.org/10.1371/journal.pmed.0030007 doi: 10.1371/journal.pmed.0030007
|
| [27] | H. L. Smith, Monotone Dynamical Systems: An Introduction to the Theory of Competitive and Cooperative Systems, American Mathematical Society, Providence, 1995. https://doi.org/10.1090/surv/041 |
| [28] |
H. L. Smith, X. Q. Zhao, Robust persistence for semidynamical systems, Nonlinear Anal., 47 (2001), 6169–6179. https://doi.org/10.1016/S0362-546X(01)00678-2 doi: 10.1016/S0362-546X(01)00678-2
|
| [29] |
Y. J. Lou, X. Q. Zhao, A reaction-diffusion malaria model with incubation period in the vector population, J. Math. Biol., 62 (2011), 543–568. https://doi.org/10.1007/s00285-010-0346-8 doi: 10.1007/s00285-010-0346-8
|
| [30] |
Y. X. Wu, X. F. Zou, Dynamics and profiles of a diffusive host-pathogen system with distinct dispersal rates, J. Differ. Equations, 264 (2018), 4989–5024. https://doi.org/10.1016/j.jde.2017.12.027 doi: 10.1016/j.jde.2017.12.027
|
| [31] |
H. Y. Cheng, Y. F. Lv, R. Yuan, Long time behavior of a degenerate NPZ model with spatial heterogeneity, Appl. Math. Lett., 132 (2022), 108088. https://doi.org/10.1016/j.aml.2022.108088 doi: 10.1016/j.aml.2022.108088
|
| [32] |
S. B. Hsu, F. B. Wang, X. Q. Zhao, Dynamics of a periodically pulsed bio-reactor model with a hydraulic storage zone, J. Dyn. Differ. Equations, 23 (2011), 817–842. https://doi.org/10.1007/s10884-011-9224-3 doi: 10.1007/s10884-011-9224-3
|
| [33] | G. Sell, Y. You, Dynamics of Evolutionary Equations, Springer, New York, 2002. https://doi.org/10.1007/978-1-4757-5037-9 |
| [34] |
P. Magal, X. Q. Zhao, Global attractors and steady states for uniformly persistent dynamical systems, SIAM J. Math. Anal., 37 (2005), 251–275. https://doi.org/10.1137/S0036141003439173 doi: 10.1137/S0036141003439173
|
| [35] | X. Q. Zhao, Dynamics Systems in Population Biology, Spring-Verlag, New York, 2017. https://doi.org/10.1007/978-0-387-21761-1 |
| [36] |
W. D. Wang, X. Q. Zhao, Basic reproduction numbers for reaction-diffusion epidemic models, SIAM. J. Appl. Dyn. Syst., 11 (2012), 1652–1673. https://doi.org/10.1137/120872942 doi: 10.1137/120872942
|
| [37] |
H. R. Thieme, Spectral bound and reproduction number for infinite-dimensional population structure and time heterogeneity, SIAM J. Appl. Math., 70 (2009), 188–211. https://doi.org/10.1137/080732870 doi: 10.1137/080732870
|
| [38] | K. J. Engel, R. Nagel, One-Parameter Semigroups for Linear Evolution Equations, Springer, New York, 1999. https://doi.org/10.1007/b97696 |
| [39] | R. D. Nussbaum, Eigenvectors of nonlinear positive operator and the linear Krein-Rutman theorem, in Fixed Point Theory, Springer, (1981), 309–331. https://doi.org/10.1007/bfb0092191 |
| [40] |
R. H. Martin, H. L. Smith, Abstract functional differential equations and reaction-diffusion systems, T. Am. Math. Soc., 321 (1990), 1–44. https://doi.org/10.1090/s0002-9947-1990-0967316-x doi: 10.1090/s0002-9947-1990-0967316-x
|
| [41] |
Y. Jin, F. B. Wang, Dynamics of a benthic-drift model for two competitive species, J. Math. Anal. Appl., 462 (2018), 840–860. https://doi.org/10.1016/j.jmaa.2017.12.050 doi: 10.1016/j.jmaa.2017.12.050
|
| [42] | W. M. Ni, The Mathematics of Diffusion, Society for Industrial and Applied Mathematics, Philadelphia, 2011. https://doi.org/10.1137/1.9781611971972 |
| [43] |
J. P. Shi, X. F. Wang, On global bifurcation for quasilinear elliptic systems on bounded domains, J. Differ. Equations, 246 (2009), 2788–2812. https://doi.org/10.1016/j.jde.2008.09.009 doi: 10.1016/j.jde.2008.09.009
|
| [44] |
M. G. Crandall, P. H. Rabinowitz, Bifurcation from simple eigenvalues, J. Funct. Anal., 8 (1971), 321–340. https://doi.org/10.1016/0022-1236(71)90015-2 doi: 10.1016/0022-1236(71)90015-2
|
| [45] |
J. P. Shi, Persistence and bifurcation of degenerate solutions, J. Funct. Anal. 69 (1999), 494–531. https://doi.org/10.1006/jfan.1999.3483 doi: 10.1006/jfan.1999.3483
|
| [46] |
H. Y. Shu, Z. M. Ma, X. S. Wang, Threshold dynamics of a nonlocal and delayed cholera model in a spatially heterogeneous environment, J. Math. Biol., 83 (2021), 1–33. https://doi.org/10.1007/s00285-021-01672-5 doi: 10.1007/s00285-021-01672-5
|
| [47] |
J. X. Xu, J. L. Wang, Threshold-type result for a nonlocal diffusive cholera model with seasonally forced intrinsic incubation period, Discrete Cont. Dyn.-B, 28 (2023), 3393–3413. https://doi.org/10.3934/dcdsb.2022223 doi: 10.3934/dcdsb.2022223
|
| [48] |
Y. H. Grad, J. C. Miller, M. Lipsitch, Cholera modeling: challenges to quantitative analysis and predicting the impact of interventions, Epidemiology, 23 (2012), 523–530. https://doi.org/10.1097/EDE.0b013e3182572581 doi: 10.1097/EDE.0b013e3182572581
|
| [49] |
X. N. Wang, H. Wang, M. Y. Li, $\mathcal{R}_{0}$ and sensitivity analysis of a predator-prey model with seasonality and maturation delay, Math. Biosci., 315 (2019), 108225. https://doi.org/10.1016/j.mbs.2019.108225 doi: 10.1016/j.mbs.2019.108225
|






Figures(7)

Shengfu Wang, Linfei Nie. Global analysis of a diffusive Cholera model with multiple transmission pathways, general incidence and incomplete immunity in a heterogeneous environment[J]. Mathematical Biosciences and Engineering, 2024, 21(4): 4927-4955. doi: 10.3934/mbe.2024218







 DownLoad:
DownLoad: