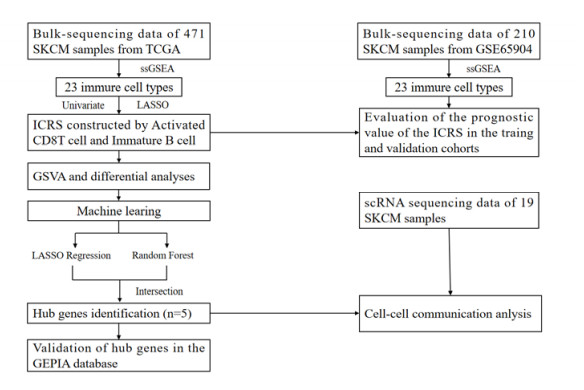
The tumor microenvironment plays a crucial role in melanoma. In this study, the abundance of immune cells in melanoma samples was assessed and analyzed using single sample gene set enrichment analysis (ssGSEA), and the predictive value of immune cells was assessed using univariate COX regression analysis. The Least Absolute Shrinkage and Selection Operator (LASSO)-Cox regression analysis was applied to construct an immune cell risk score (ICRS) model with a high predictive value for identifying the immune profile of melanoma patients. The pathway enrichment between the different ICRS groups was also elucidated. Next, five hub genes for diagnosing the prognosis of melanoma were screened by two machine learning algorithms, LASSO and random forest. The distribution of hub genes in immune cells was analyzed on account of Single-cell RNA sequencing (scRNA-seq), and the interaction between genes and immune cells was elucidated by cellular communication. Ultimately, the ICRS model on account of two types of immune cells (Activated CD8 T cell and Immature B cell) was constructed and validated, which can determine melanoma prognosis. In addition, five hub genes were identified as potential therapeutic targets affecting the prognosis of melanoma patients.
Citation: Linqian Guo, Qingrong Meng, Wenqi Lin, Kaiyuan Weng. Identification of immune subtypes of melanoma based on single-cell and bulk RNA sequencing data[J]. Mathematical Biosciences and Engineering, 2023, 20(2): 2920-2936. doi: 10.3934/mbe.2023138
The tumor microenvironment plays a crucial role in melanoma. In this study, the abundance of immune cells in melanoma samples was assessed and analyzed using single sample gene set enrichment analysis (ssGSEA), and the predictive value of immune cells was assessed using univariate COX regression analysis. The Least Absolute Shrinkage and Selection Operator (LASSO)-Cox regression analysis was applied to construct an immune cell risk score (ICRS) model with a high predictive value for identifying the immune profile of melanoma patients. The pathway enrichment between the different ICRS groups was also elucidated. Next, five hub genes for diagnosing the prognosis of melanoma were screened by two machine learning algorithms, LASSO and random forest. The distribution of hub genes in immune cells was analyzed on account of Single-cell RNA sequencing (scRNA-seq), and the interaction between genes and immune cells was elucidated by cellular communication. Ultimately, the ICRS model on account of two types of immune cells (Activated CD8 T cell and Immature B cell) was constructed and validated, which can determine melanoma prognosis. In addition, five hub genes were identified as potential therapeutic targets affecting the prognosis of melanoma patients.

| [1] |
T. Yamauchi, S. Shangraw, Z. Zhai, D. Ravindran Menon, N. Batta, R. P. Dellavalle, et al., Alcohol as a non-UV social-environmental risk factor for melanoma, Cancers (Basel), 14 (2022), 5010. https://doi:10.3390/cancers14205010 doi: 10.3390/cancers14205010

|
| [2] |
F. S. Hodi, S. J. O'Day, D. F. McDermott, R. W. Weber, J. A. Sosman, J. B. Haanen, et al., Improved survival with ipilimumab in patients with metastatic melanoma, N. Engl. J. Med., 363 (2010), 711–723. https://doi:10.1056/NEJMoa1003466 doi: 10.1056/NEJMoa1003466
|
| [3] |
Y. Qu, S. Zhang, Y. Zhang, X. Feng, F. Wang, Identification of immune-related genes with prognostic significance in the microenvironment of cutaneous melanoma, Virchows Arch., 478 (2021), 943–959. https://doi:10.1007/s00428-020-02948-9 doi: 10.1007/s00428-020-02948-9
|
| [4] |
Y. Xie, J. Zhang, M. Li, Y. Zhang, Q. Li, Y. Zheng, et al., Identification of lactate-related gene signature for prediction of progression and immunotherapeutic response in skin cutaneous melanoma, Front. Oncol., 12 (2022), 818868. https://doi:10.3389/fonc.2022.818868 doi: 10.3389/fonc.2022.818868
|
| [5] |
N. A. N. Jorge, J. G. V. Cruz, M. A. M. Pretti, M. H. Bonamino, P. A. Possik, M. Boroni, Poor clinical outcome in metastatic melanoma is associated with a microRNA-modulated immunosuppressive tumor microenvironment, J. Transl. Med., 18 (2020), 56. https://doi:10.1186/s12967-020-02235-w doi: 10.1186/s12967-020-02235-w
|
| [6] |
L. Fattore, C. F. Ruggiero, D. Liguoro, R. Mancini, G. Ciliberto, Single cell analysis to dissect molecular heterogeneity and disease evolution in metastatic melanoma, Cell Death Dis., 10 (2019), 827. https://doi:10.1038/s41419-019-2048-5 doi: 10.1038/s41419-019-2048-5
|
| [7] |
A. J. Combes, B. Samad, J. Tsui, N. W. Chew, P. Yan, G. C. Reeder, et al., Discovering dominant tumor immune archetypes in a pan-cancer census, Cell, 185 (2022), 184–203. https://doi:10.1016/j.cell.2021.12.004 doi: 10.1016/j.cell.2021.12.004
|
| [8] |
S. S. Potter, Single-cell RNA sequencing for the study of development, physiology and disease, Nat. Rev. Nephrol., 14 (2018), 479–492. https://doi:10.1038/s41581-018-0021-7 doi: 10.1038/s41581-018-0021-7
|
| [9] |
K. Asada, K. Takasawa, H. Machino, S. Takahashi, N. Shinkai, A. Bolatkan, et al., Single-cell analysis using machine learning techniques and its application to medical research, Biomedicines, 9 (2021), 1513. https://doi:10.3390/biomedicines9111513 doi: 10.3390/biomedicines9111513
|
| [10] |
M. P. Kumar, J. Du, G. Lagoudas, Y. Jiao, A. Sawyer, D. C. Drummond, et al., Analysis of single-cell RNA-Seq identifies cell-cell communication associated with tumor characteristics, Cell Rep., 25 (2018), 1458–1468. https://doi:10.1016/j.celrep.2018.10.047 doi: 10.1016/j.celrep.2018.10.047
|
| [11] |
J. Chen, S. Suo, P. P. Tam, J. J. Han, G. Peng, N. Jing, Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq, Nat. Protoc., 12 (2017), 566–580. https://doi:10.1038/nprot.2017.003 doi: 10.1038/nprot.2017.003
|
| [12] |
L. Huang, C. Wu, D. Xu, Y. Cui, J. Tang, Screening of important factors in the early sepsis stage based on the evaluation of ssGSEA algorithm and ceRNA regulatory network, Evol. Bioinf. Online, 17 (2021), 11769343211058463. https://doi:10.1177/11769343211058463 doi: 10.1177/11769343211058463
|
| [13] |
Y. Li, J. Liu, X. Gao, B. Jie, M. Kim, P. T. Yap, et al., Multimodal hyper-connectivity of functional networks using functionally-weighted LASSO for MCI classification, Med. Image Anal., 52 (2019), 80–96. https://doi:10.1016/j.media.2018.11.006 doi: 10.1016/j.media.2018.11.006
|
| [14] |
K. Yoshihara, M. Shahmoradgoli, E. Martínez, R. Vegesna, H. Kim, W. Torres-Garcia, et al., Inferring tumour purity and stromal and immune cell admixture from expression data, Nat. Commun., 4 (2013), 2612. https://doi:10.1038/ncomms3612 doi: 10.1038/ncomms3612
|
| [15] |
X. Deng, T. Li, L. Mo, F. Wang, J. Ji, X. He, et al., Machine learning model for the prediction of prostate cancer in patients with low prostate-specific antigen levels: A multicenter retrospective analysis, Front. Oncol., 12 (2022), 985940. https://doi:10.3389/fonc.2022.985940 doi: 10.3389/fonc.2022.985940
|
| [16] |
Y. Zhou, W. Shi, D. Zhao, S. Xiao, K. Wang, J. Wang, Identification of immune-associated genes in diagnosing aortic valve calcification with metabolic syndrome by integrated bioinformatics analysis and machine learning, Front. Immunol., 13 (2022), 937886. https://doi:10.3389/fimmu.2022.937886 doi: 10.3389/fimmu.2022.937886
|
| [17] |
J. Vallejo, R. Saigusa, R. Gulati, S. S. Armstrong Suthahar, V. Suryawanshi, A. Alimadadi, et al., Combined protein and transcript single-cell RNA sequencing in human peripheral blood mononuclear cells, BMC Biol., 20 (2022), 193. https://doi:10.1186/s12915-022-01382-4 doi: 10.1186/s12915-022-01382-4
|
| [18] |
M. Efremova, M. Vento-Tormo, S. A. Teichmann, R. Vento-Tormo, CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes, Nat. Protoc., 15 (2020), 1484–1506. https://doi:10.1038/s41596-020-0292-x doi: 10.1038/s41596-020-0292-x
|
| [19] |
D. Hanahan, R. A. Weinberg, Hallmarks of cancer: the next generation, Cell, 144 (2011), 646–674. https://doi:10.1016/j.cell.2011.02.013 doi: 10.1016/j.cell.2011.02.013
|
| [20] |
A. D. Therien, G. M. Beasley, K. E. Rhodin, N. E. Farrow, D. S. Tyler, D. Boczkowski, et al., Spatial biology analysis reveals B cell follicles in secondary lymphoid structures may regulate anti-tumor responses at initial melanoma diagnosis, Front. Immunol., 13 (2022), 952220. https://doi:10.3389/fimmu.2022.952220 doi: 10.3389/fimmu.2022.952220
|
| [21] |
M. S. Ryu, M. Y. Woo, D. Kwon, A. E. Hong, K. Y. Song, S. Park, et al., Accumulation of cytolytic CD8+ T cells in B16-melanoma and proliferation of mature T cells in TIS21-knockout mice after T cell receptor stimulation, Exp. Cell Res., 327 (2014), 209–221. https://doi:10.1016/j.yexcr.2014.07.028 doi: 10.1016/j.yexcr.2014.07.028
|
| [22] |
B. Song, H. Chi, G. Peng, Y. Song, Z. Cui, Y. Zhu, et al., Characterization of coagulation-related gene signature to predict prognosis and tumor immune microenvironment in skin cutaneous melanoma, Front. Oncol., 12 (2022), 975255. https://doi:10.3389/fonc.2022.975255 doi: 10.3389/fonc.2022.975255
|
| [23] |
X. Yang, X. Wang, X. Sun, M. Xiao, L. Fan, Y. Su, et al., Construction of five cuproptosis-related lncRNA signature for predicting prognosis and immune activity in skin cutaneous melanoma, Front. Genet., 13 (2022), 972899. https://doi:10.3389/fgene.2022.972899 doi: 10.3389/fgene.2022.972899
|
| [24] |
W. Fu, G. Ma, Significance of immunogenic cell death-related genes in prognosis prediction and immune microenvironment landscape of patients with cutaneous melanoma, Front. Genet., 13 (2022), 988821. https://doi:10.3389/fgene.2022.988821 doi: 10.3389/fgene.2022.988821
|
| [25] |
C. S. Groeneveld, J. Fontugne, L. Cabel, I. Bernard-Pierrot, F. Radvanyi, Y. Allory, et al., Tertiary lymphoid structures marker CXCL13 is associated with better survival for patients with advanced-stage bladder cancer treated with immunotherapy, Eur. J. Cancer, 148 (2021), 181–189. https://doi:10.1016/j.ejca.2021.01.036 doi: 10.1016/j.ejca.2021.01.036
|
| [26] |
L. Shen, J. Li, Q. Liu, M. Das, W. Song, X. Zhang, et al., Nano-trapping CXCL13 reduces regulatory B cells in tumor microenvironment and inhibits tumor growth, J. Control. Release, 343 (2022), 303–313. https://doi:10.1016/j.jconrel.2022.01.039 doi: 10.1016/j.jconrel.2022.01.039
|
| [27] |
Z. Si, H. Hu, Identification of CXCL13 as an Immune-related biomarker associated with tumorigenesis and prognosis in cutaneous melanoma patients, Med. Sci. Monit., 27 (2021), e932052. https://doi:10.12659/msm.932052 doi: 10.12659/msm.932052
|
| [28] |
F. J. Li, D. M. Schreeder, R. Li, J. Wu, R. S. Davis, FCRL3 promotes TLR9-induced B-cell activation and suppresses plasma cell differentiation, Eur. J. Immunol., 43 (2013), 2980–2992. https://doi:10.1002/eji.201243068 doi: 10.1002/eji.201243068
|
| [29] |
Y. Li, S. Lyu, Z. Gao, W. Zha, P. Wang, Y. Shan, et al., Identification of potential prognostic biomarkers associated with cancerometastasis in skin cutaneous melanoma, Front. Genet., 12 (2021), 687979. https://doi:10.3389/fgene.2021.687979 doi: 10.3389/fgene.2021.687979
|
| [30] |
D. B. Kuhns, A. P. Hsu, D. Sun, K. Lau, D. Fink, P. Griffith, et al., NCF1 (p47(phox))-deficient chronic granulomatous disease: comprehensive genetic and flow cytometric analysis, Blood Adv., 3 (2019), 136–147. https://doi:10.1182/bloodadvances.2018023184 doi: 10.1182/bloodadvances.2018023184
|
| [31] |
B. Yigit, N. Wang, E. T. Hacken, S. S. Chen, A. K. Bhan, A. Suarez-Fueyo, et al., SLAMF6 as a regulator of exhausted CD8(+) T cells in cancer, Cancer Immunol. Res., 7 (2019), 1485–1496. https://doi:10.1158/2326-6066.Cir-18-0664 doi: 10.1158/2326-6066.Cir-18-0664
|
| [32] |
B. Liu, L. Zeng, Y. Shao, R. Fu, Expression and function of SLAMF6 in CD8(+) T lymphocytes of patients with severe aplastic anemia, Cell Immunol., 364 (2021), 104343. https://doi:10.1016/j.cellimm.2021.104343 doi: 10.1016/j.cellimm.2021.104343
|
| [33] |
E. Hajaj, G. Eisenberg, S. Klein, S. Frankenburg, S. Merims, I. B. David, et al., SLAMF6 deficiency augments tumor killing and skews toward an effector phenotype revealing it as a novel T cell checkpoint, Elife, 9 (2020). https://doi:10.7554/eLife.52539 doi: 10.7554/eLife.52539
|
| [34] |
I. Tirosh, B. Izar, S. M. Prakadan, M. H. Wadsworth, D. Treacy, J. J. Trombetta, et al., Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq, Science, 352 (2016), 189–196. https://doi:10.1126/science.aad0501 doi: 10.1126/science.aad0501
|
| [35] |
J. Griss, W. Bauer, C. Wagner, M. Simon, M. Chen, K. Grabmeier-Pfistershammer, et al., B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma, Nat. Commun., 10 (2019), 4186. https://doi:10.1038/s41467-019-12160-2 doi: 10.1038/s41467-019-12160-2
|
| [36] |
W. H. Fridman, F. Petitprez, M. Meylan, T. W. Chen, C. M. Sun, L. T. Roumenina, et al., B cells and cancer: To B or not to B, J. Exp. Med., 218 (2021), e20200851. https://doi:10.1084/jem.20200851 doi: 10.1084/jem.20200851
|
| [37] |
M. Lauss, M. Donia, I. M. Svane, G. Jönsson, B cells and tertiary lymphoid structures: Friends or foes in cancer immunotherapy, Clin. Cancer Res., 28 (2022), 1751–1758. https://doi:10.1158/1078-0432.Ccr-21-1130 doi: 10.1158/1078-0432.Ccr-21-1130
|
| [38] |
T. Gambichler, M. Bindsteiner, S. Höxtermann, S. Terras, A. Kreuter, Circulating CD4+ CD25 (high) CD127 (low) regulatory T cells are an independent predictor of advanced melanoma, Pigment Cell Melanoma Res., 26 (2013), 280–283. https://doi:10.1111/pcmr.12055 doi: 10.1111/pcmr.12055
|
| [39] |
C. Miracco, V. Mourmouras, M. Biagioli, P. Rubegni, S. Mannucci, I. Monciatti, et al., Utility of tumour-infiltrating CD25+FOXP3+ regulatory T cell evaluation in predicting local recurrence in vertical growth phase cutaneous melanoma, Oncol. Rep., 18 (2007), 1115–1122. https://doi.org/10.3892/or.18.5.1115 doi: 10.3892/or.18.5.1115
|
| [40] |
P. A. C. Costa, W. N. Silva, P. Prazeres, C. C. Picoli, G. D. A. Guardia, A. C. Costa, et al., Chemogenetic modulation of sensory neurons reveals their regulating role in melanoma progression, Acta Neuropathol. Commun., 9 (2021), 183. https://doi:10.1186/s40478-021-01273-9 doi: 10.1186/s40478-021-01273-9
|
| [41] |
S. Kalaora, A. Nagler, J. A. Wargo, Y. Samuels, Mechanisms of immune activation and regulation: lessons from melanoma, Nat. Rev. Cancer, 22 (2022), 195–207. https://doi:10.1038/s41568-022-00442-9 doi: 10.1038/s41568-022-00442-9
|
| [42] |
M. Hussain, D. Adah, M. Tariq, Y. Lu, J. Zhang, J. Liu, CXCL13/CXCR5 signaling axis in cancer, Life Sci., 227 (2019), 175–186. https://doi:10.1016/j.lfs.2019.04.053 doi: 10.1016/j.lfs.2019.04.053
|
| [43] |
A. Cagigi, F. Mowafi, L. V. Phuong Dang, K. Tenner-Racz, A. Atlas, S. Grutzmeier, et al., Altered expression of the receptor-ligand pair CXCR5/CXCL13 in B cells during chronic HIV-1 infection, Blood, 112 (2008), 4401–4410. https://doi.org/10.1182/blood-2008-02-140426 doi: 10.1182/blood-2008-02-140426
|







Figures(8)

Linqian Guo, Qingrong Meng, Wenqi Lin, Kaiyuan Weng. Identification of immune subtypes of melanoma based on single-cell and bulk RNA sequencing data[J]. Mathematical Biosciences and Engineering, 2023, 20(2): 2920-2936. doi: 10.3934/mbe.2023138







 DownLoad:
DownLoad: