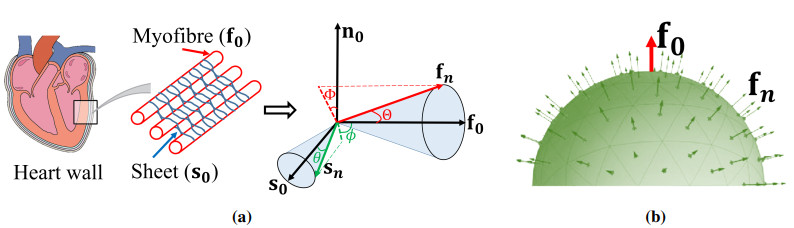
It is widely acknowledged that an imbalanced biomechanical environment can have significant effects on myocardial pathology, leading to adverse remodelling of cardiac function if it persists. Accurate stress prediction essentially depends on the strain energy function which should have competent descriptive and predictive capabilities. Previous studies have focused on myofibre dispersion, but not on fibres along other directions. In this study, we will investigate how fibre dispersion affects myocardial biomechanical behaviours by taking into account both the myofibre dispersion and the sheet fibre dispersion, with a focus on the sheet fibre dispersion. Fibre dispersion is incorporated into a widely-used myocardial strain energy function using the discrete fibre bundle approach. We first study how different dispersion affects the descriptive capability of the strain energy function when fitting to ex vivo experimental data, and then the predictive capability in a human left ventricle during diastole. Our results show that the chosen strain energy function can achieve the best goodness-of-fit to the experimental data by including both fibre dispersion. Furthermore, noticeable differences in stress can be found in the LV model. Our results may suggest that it is necessary to include both dispersion for myofibres and the sheet fibres for the improved descriptive capability to the ex vivo experimental data and potentially more accurate stress prediction in cardiac mechanics.
Citation: Debao Guan, Yuqian Mei, Lijian Xu, Li Cai, Xiaoyu Luo, Hao Gao. Effects of dispersed fibres in myocardial mechanics, Part I: passive response[J]. Mathematical Biosciences and Engineering, 2022, 19(4): 3972-3993. doi: 10.3934/mbe.2022183
It is widely acknowledged that an imbalanced biomechanical environment can have significant effects on myocardial pathology, leading to adverse remodelling of cardiac function if it persists. Accurate stress prediction essentially depends on the strain energy function which should have competent descriptive and predictive capabilities. Previous studies have focused on myofibre dispersion, but not on fibres along other directions. In this study, we will investigate how fibre dispersion affects myocardial biomechanical behaviours by taking into account both the myofibre dispersion and the sheet fibre dispersion, with a focus on the sheet fibre dispersion. Fibre dispersion is incorporated into a widely-used myocardial strain energy function using the discrete fibre bundle approach. We first study how different dispersion affects the descriptive capability of the strain energy function when fitting to ex vivo experimental data, and then the predictive capability in a human left ventricle during diastole. Our results show that the chosen strain energy function can achieve the best goodness-of-fit to the experimental data by including both fibre dispersion. Furthermore, noticeable differences in stress can be found in the LV model. Our results may suggest that it is necessary to include both dispersion for myofibres and the sheet fibres for the improved descriptive capability to the ex vivo experimental data and potentially more accurate stress prediction in cardiac mechanics.

| [1] |
M. R. Zile, C. F. Baicu, W. H. Gaasch, Diastolic heart failure—abnormalities in active relaxation and passive stiffness of the left ventricle, N. Engl. J. Med., 350 (2004), 1953–1959. https://doi.org/10.1056/NEJMoa032566 doi: 10.1056/NEJMoa032566

|
| [2] |
K. Mangion, H. Gao, D. Husmeier, X. Luo, C. Berry, Advances in computational modelling for personalised medicine after myocardial infarction, Heart, 104 (2018), 550–557. http://dx.doi.org/10.1136/heartjnl-2017-311449 doi: 10.1136/heartjnl-2017-311449
|
| [3] |
G. A. Holzapfel, R. W. Ogden, Constitutive modelling of passive myocardium: a structurally based framework for material characterization, Philos. Trans. R. Soc., A, 367 (2009), 3445–3475. https://doi.org/10.1098/rsta.2009.0091 doi: 10.1098/rsta.2009.0091
|
| [4] |
H. Gao, H. Wang, C. Berry, X. Luo, B. E. Griffith, Quasi-static image-based immersed boundary-finite element model of left ventricle under diastolic loading, Int. J. Numer. Method. Biomed. Eng., 30 (2014), 1199–1222. https://doi.org/10.1002/cnm.2652 doi: 10.1002/cnm.2652
|
| [5] |
H. Gao, A. Aderhold, K. Mangion, X. Luo, D. Husmeier, C. Berry, Changes and classification in myocardial contractile function in the left ventricle following acute myocardial infarction, J. R. Soc. Interface, 14 (2017), 20170203. https://doi.org/10.1098/rsif.2017.0203 doi: 10.1098/rsif.2017.0203
|
| [6] |
B. Baillargeon, N. Rebelo, D. D. Fox, R. L. Taylor, E. Kuhl, The living heart project: a robust and integrative simulator for human heart function, Eur. J. Mech. A/Solids, 48 (2014), 38–47. https://doi.org/10.1016/j.euromechsol.2014.04.001 doi: 10.1016/j.euromechsol.2014.04.001
|
| [7] | K. L. Sack, E. Aliotta, D. B. Ennis, J. S. Choy, G. S. Kassab, J. M. Guccione, et al., Construction and validation of subject-specific biventricular finite-element models of healthy and failing swine hearts from high-resolution dt-mri, Front. Physiol., 9 (2018). https://doi.org/10.3389/fphys.2018.00539 |
| [8] |
H. Gao, K. Mangion, D. Carrick, D. Husmeier, X. Luo, C. Berry, Estimating prognosis in patients with acute myocardial infarction using personalized computational heart models, Sci. Rep., 7 (2017), 13527. https://doi.org/10.1038/s41598-017-13635-2 doi: 10.1038/s41598-017-13635-2
|
| [9] |
S. I. H. Richardson, H. Gao, J. Cox, R. Janiczek, B. E. Griffith, C. Berry, et al., A poroelastic immersed finite element framework for modelling cardiac perfusion and fluid–structure interaction, Int. J. Numer. Method. Biomed. Eng., 37 (2021), e3446. https://doi.org/10.1002/cnm.3446 doi: 10.1002/cnm.3446
|
| [10] |
H. M. Wang, H. Gao, X. Y. Luo, C. Berry, B. E. Griffith, R. W. Ogden, et al., Structure-based finite strain modelling of the human left ventricle in diastole, Int. J. Numer. Method. Biomed. Eng., 29 (2013), 83–103. https://doi.org/10.1002/cnm.2497 doi: 10.1002/cnm.2497
|
| [11] | D. Guan, X. Luo, H. Gao, Constitutive modelling of soft biological tissue from ex vivo to in vivo: myocardium as an example, in International Conference by Center for Mathematical Modeling and Data Science, Osaka University, Springer, (2020), 3–14. https://doi.org/10.1007/978-981-16-4866-3_1 |
| [12] |
H. Gao, W. G. Li, L. Cai, C. Berry, X. Y. Luo, Parameter estimation in a holzapfel–ogden law for healthy myocardium, J. Eng. Math., 95 (2015), 231–248. https://doi.org/10.1007/s10665-014-9740-3 doi: 10.1007/s10665-014-9740-3
|
| [13] |
U. Noè, A. Lazarus, H. Gao, V. Davies, B. Macdonald, K. Mangion, et al., Gaussian process emulation to accelerate parameter estimation in a mechanical model of the left ventricle: a critical step towards clinical end-user relevance, J. R. Soc. Interface, 16 (2019), 20190114. https://doi.org/10.1098/rsif.2019.0114 doi: 10.1098/rsif.2019.0114
|
| [14] |
V. Davies, U. Noè, A. Lazarus, H. Gao, B. Macdonald, C. Berry, et al., Fast parameter inference in a biomechanical model of the left ventricle by using statistical emulation, J. R. Stat. Soc. Ser. C Appl. Stat., 68 (2019), 1555–1576. https://doi.org/10.1111/rssc.12374 doi: 10.1111/rssc.12374
|
| [15] |
G. Sommer, A. J. Schriefl, M. Andrä, M. Sacherer, C. Viertler, H. Wolinski, et al., Biomechanical properties and microstructure of human ventricular myocardium, Acta Biomater., 24 (2015), 172–192. https://doi.org/10.1016/j.actbio.2015.06.031 doi: 10.1016/j.actbio.2015.06.031
|
| [16] |
F. Ahmad, S. Soe, N. White, R. Johnston, I. Khan, J. Liao, et al., Region-specific microstructure in the neonatal ventricles of a porcine model, Ann. Biomed. Eng., 46 (2018), 2162–2176. https://doi.org/10.1007/s10439-018-2089-4 doi: 10.1007/s10439-018-2089-4
|
| [17] |
Y. Lanir, Multi-scale structural modeling of soft tissues mechanics and mechanobiology, J. Elast., 129 (2017), 7–48. https://doi.org/10.1007/s10659-016-9607-0 doi: 10.1007/s10659-016-9607-0
|
| [18] |
T. C. Gasser, R. W. Ogden, G. A. Holzapfel, Hyperelastic modelling of arterial layers with distributed collagen fibre orientations, J. R. Soc. Interface, 3 (2006), 15–35. https://doi.org/10.1098/rsif.2005.0073 doi: 10.1098/rsif.2005.0073
|
| [19] |
T. S. E. Eriksson, A. J. Prassl, G. Plank, G. A. Holzapfel, Modeling the dispersion in electromechanically coupled myocardium, Int. J. Numer. Method. Biomed. Eng., 29 (2013), 1267–1284. https://doi.org/10.1002/cnm.2575 doi: 10.1002/cnm.2575
|
| [20] | G. A. Holzapfel, R. W. Ogden, S. Sherifova, On fibre dispersion modelling of soft biological tissues: a review, Proc. Math. Phys. Eng. Sci., 475 (2019). https://doi.org/10.1098/rspa.2018.0736 |
| [21] |
A. V. Melnik, X. Luo, R. W. Ogden, A generalised structure tensor model for the mixed invariant i8, Int. J. Non-Linear Mech., 107 (2018), 137–148. https://doi.org/10.1016/j.ijnonlinmec.2018.08.018 doi: 10.1016/j.ijnonlinmec.2018.08.018
|
| [22] |
A. Pandolfi, A. Gizzi, M. Vasta, Coupled electro-mechanical models of fiber-distributed active tissues, J. Biomech., 49 (2016), 2436–2444. https://doi.org/10.1016/j.jbiomech.2016.01.038 doi: 10.1016/j.jbiomech.2016.01.038
|
| [23] |
A. Gizzi, A. Pandolfi, M. Vasta, Statistical characterization of the anisotropic strain energy in soft materials with distributed fibers, Mech. Mater., 92 (2016), 119–138. https://doi.org/10.1016/j.mechmat.2015.09.008 doi: 10.1016/j.mechmat.2015.09.008
|
| [24] |
D. Guan, X. Zhuan, W. Holmes, X. Luo, H. Gao, Modelling of fibre dispersion and its effects on cardiac mechanics from diastole to systole, J. Eng. Math., 128 (2021), 1–24. https://doi.org/10.1007/s10665-021-10102-w doi: 10.1007/s10665-021-10102-w
|
| [25] |
G. A. Holzapfel, R. W. Ogden, On fiber dispersion models: exclusion of compressed fibers and spurious model comparisons, J. Elast., 129 (2017), 49–68. https://doi.org/10.1007/s10659-016-9605-2 doi: 10.1007/s10659-016-9605-2
|
| [26] |
K. Li, R. W. Ogden, G. A. Holzapfel, A discrete fibre dispersion method for excluding fibres under compression in the modelling of fibrous tissues, J. R. Soc. Interface, 15 (2018), 20170766. https://doi.org/10.1098/rsif.2017.0766 doi: 10.1098/rsif.2017.0766
|
| [27] |
D. Guan, J. Yao, X. Luo, H. Gao, Effect of myofibre architecture on ventricular pump function by using a neonatal porcine heart model: from dt-mri to rule-based methods, R. Soc. Open Sci., 7 (2020), 191655. https://doi.org/10.1098/rsos.191655 doi: 10.1098/rsos.191655
|
| [28] |
K. Li, R. W. Ogden, G. A. Holzapfel, Computational method for excluding fibers under compression in modeling soft fibrous solids, Eur. J. Mech. A/Solids, 57 (2016), 178–193. https://doi.org/10.1016/j.euromechsol.2015.11.003 doi: 10.1016/j.euromechsol.2015.11.003
|
| [29] |
M. Vasta, A. Gizzi, A. Pandolfi, On three-and two-dimensional fiber distributed models of biological tissues, Probab. Eng. Mech., 37 (2014), 170–179. https://doi.org/10.1016/j.probengmech.2014.05.003 doi: 10.1016/j.probengmech.2014.05.003
|
| [30] |
G. A. Holzapfel, J. A. Niestrawska, R. W. Ogden, A. J. Reinisch, A. J. Schriefl, Modelling non-symmetric collagen fibre dispersion in arterial walls, J. R. Soc. Interface, 12 (2015), 20150188. https://doi.org/10.1098/rsif.2015.0188 doi: 10.1098/rsif.2015.0188
|
| [31] |
F. Ahmad, J. Liao, S. Soe, M. D. Jones, J. Miller, P. Berthelson, et al., Biomechanical properties and microstructure of neonatal porcine ventricles, J. Mech. Behav. Biomed. Mater., 88 (2018), 18–28. https://doi.org/10.1016/j.jmbbm.2018.07.038 doi: 10.1016/j.jmbbm.2018.07.038
|
| [32] |
G. A. Holzapfel, T. C. Gasser, R. W. Ogden, Comparison of a multi-layer structural model for arterial walls with a fung-type model, and issues of material stability, J. Biomech. Eng., 126 (2004), 264–275. https://doi.org/10.1115/1.1695572 doi: 10.1115/1.1695572
|
| [33] |
D. Guan, F. Ahmad, P. Theobald, S. Soe, X. Luo, H. Gao, On the aic-based model reduction for the general holzapfel–ogden myocardial constitutive law, Biomech. Model. Mechanobiol., 18 (2019), 1213–1232. https://doi.org/10.1007/s10237-019-01140-6 doi: 10.1007/s10237-019-01140-6
|
| [34] |
S. Klotz, I. Hay, M. L. Dickstein, G. Yi, J. Wang, M. S. Maurer, et al., Single-beat estimation of end-diastolic pressure-volume relationship: a novel method with potential for noninvasive application, Am. J. Physiol. Heart Circ. Physiol., 291 (2006), H403–H412. https://doi.org/10.1152/ajpheart.01240.2005 doi: 10.1152/ajpheart.01240.2005
|
| [35] |
A. V. Melnik, H. B. Da Rocha, A. Goriely, On the modeling of fiber dispersion in fiber-reinforced elastic materials, Int. J. Non-Linear Mech., 75 (2015), 92–106. https://doi.org/10.1016/j.ijnonlinmec.2014.10.006 doi: 10.1016/j.ijnonlinmec.2014.10.006
|
| [36] |
D. H. Cortes, S. P. Lake, J. A. Kadlowec, L. J. Soslowsky, D. M. Elliott, Characterizing the mechanical contribution of fiber angular distribution in connective tissue: comparison of two modeling approaches, Biomech. Model. Mechanobiol., 9 (2010), 651–658. https://doi.org/10.1007/s10237-010-0194-x doi: 10.1007/s10237-010-0194-x
|
| [37] |
X. Zhuan, X. Luo, H. Gao, R. W. Ogden, Coupled agent-based and hyperelastic modelling of the left ventricle post-myocardial infarction, Int. J. Numer. Method. Biomed. Eng., 35 (2019), e3155. https://doi.org/10.1002/cnm.3155 doi: 10.1002/cnm.3155
|
| [38] |
G. A. Holzapfel, R. W. Ogden, An arterial constitutive model accounting for collagen content and cross-linking, J. Mech. Phys. Solids, 136 (2020), 103682. https://doi.org/10.1016/j.jmps.2019.103682 doi: 10.1016/j.jmps.2019.103682
|
| [39] |
J. Xi, P. Lamata, S. Niederer, S. Land, W. Shi, X. Zhuang, et al., The estimation of patient-specific cardiac diastolic functions from clinical measurements, Med. Image Anal., 17 (2013), 133–146. https://doi.org/10.1016/j.media.2012.08.001 doi: 10.1016/j.media.2012.08.001
|
| [40] |
M. Strocchi, M. A. F. Gsell, C. M. Augustin, O. Razeghi, C. H. Roney, A. J. Prassl, et al., Simulating ventricular systolic motion in a four-chamber heart model with spatially varying robin boundary conditions to model the effect of the pericardium, J. Biomech., 101 (2020), 109645. https://doi.org/10.1016/j.jbiomech.2020.109645 doi: 10.1016/j.jbiomech.2020.109645
|








Figures(9) / Tables(5)

Debao Guan, Yuqian Mei, Lijian Xu, Li Cai, Xiaoyu Luo, Hao Gao. Effects of dispersed fibres in myocardial mechanics, Part I: passive response[J]. Mathematical Biosciences and Engineering, 2022, 19(4): 3972-3993. doi: 10.3934/mbe.2022183







 DownLoad:
DownLoad: