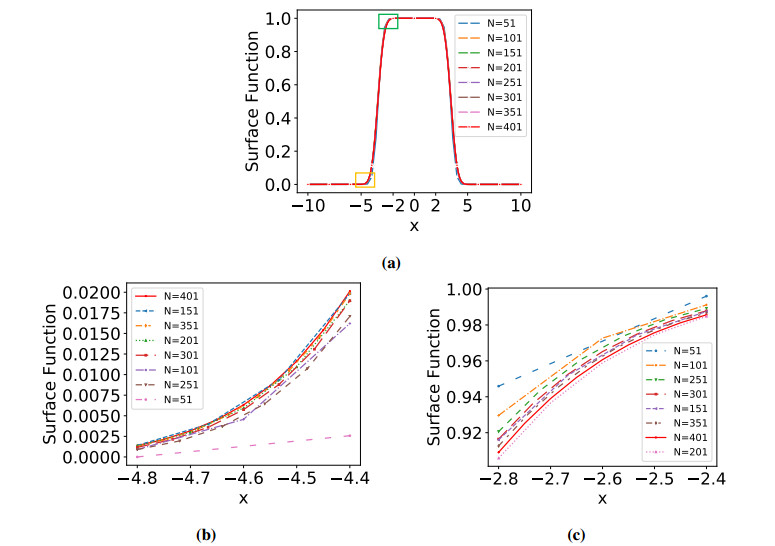
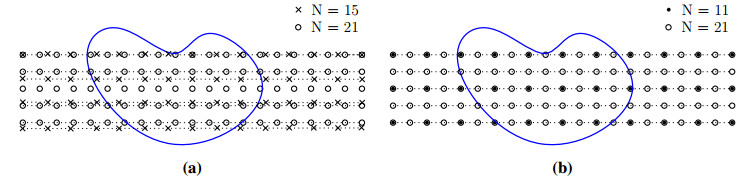
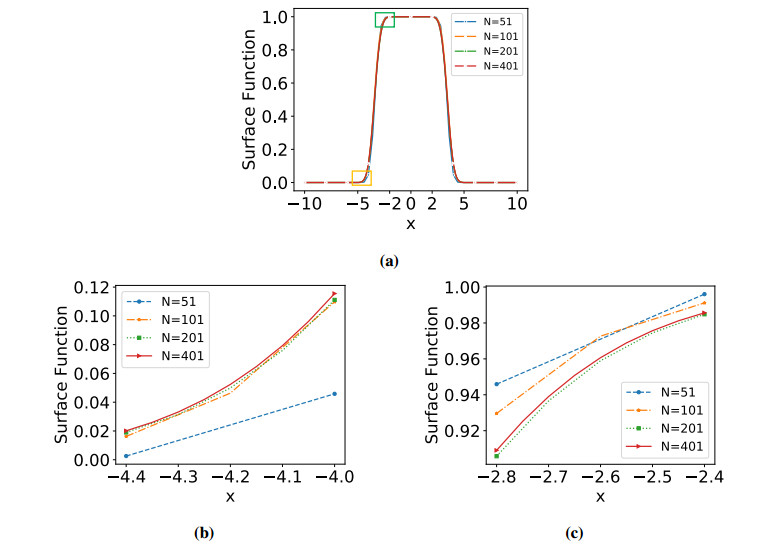
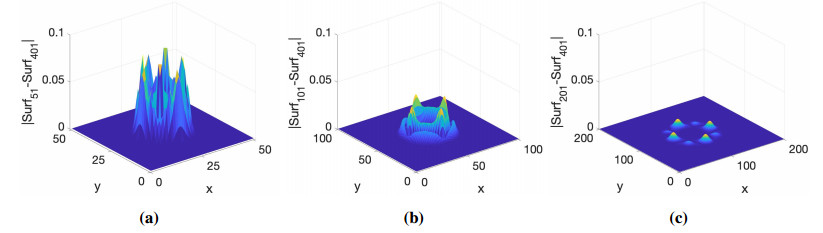
Numerical treatment of singular charges is a grand challenge in solving the Poisson-Boltzmann (PB) equation for analyzing electrostatic interactions between the solute biomolecules and the surrounding solvent with ions. For diffuse interface PB models in which solute and solvent are separated by a smooth boundary, no effective algorithm for singular charges has been developed, because the fundamental solution with a space dependent dielectric function is intractable. In this work, a novel regularization formulation is proposed to capture the singularity analytically, which is the first of its kind for diffuse interface PB models. The success lies in a dual decomposition – besides decomposing the potential into Coulomb and reaction field components, the dielectric function is also split into a constant base plus space changing part. Using the constant dielectric base, the Coulomb potential is represented analytically via Green's functions. After removing the singularity, the reaction field potential satisfies a regularized PB equation with a smooth source. To validate the proposed regularization, a Gaussian convolution surface (GCS) is also introduced, which efficiently generates a diffuse interface for three-dimensional realistic biomolecules. The performance of the proposed regularization is examined by considering both analytical and GCS diffuse interfaces, and compared with the trilinear method. Moreover, the proposed GCS-regularization algorithm is validated by calculating electrostatic free energies for a set of proteins and by estimating salt affinities for seven protein complexes. The results are consistent with experimental data and estimates of sharp interface PB models.
Citation: Siwen Wang, Emil Alexov, Shan Zhao. On regularization of charge singularities in solving the Poisson-Boltzmann equation with a smooth solute-solvent boundary[J]. Mathematical Biosciences and Engineering, 2021, 18(2): 1370-1405. doi: 10.3934/mbe.2021072
Numerical treatment of singular charges is a grand challenge in solving the Poisson-Boltzmann (PB) equation for analyzing electrostatic interactions between the solute biomolecules and the surrounding solvent with ions. For diffuse interface PB models in which solute and solvent are separated by a smooth boundary, no effective algorithm for singular charges has been developed, because the fundamental solution with a space dependent dielectric function is intractable. In this work, a novel regularization formulation is proposed to capture the singularity analytically, which is the first of its kind for diffuse interface PB models. The success lies in a dual decomposition – besides decomposing the potential into Coulomb and reaction field components, the dielectric function is also split into a constant base plus space changing part. Using the constant dielectric base, the Coulomb potential is represented analytically via Green's functions. After removing the singularity, the reaction field potential satisfies a regularized PB equation with a smooth source. To validate the proposed regularization, a Gaussian convolution surface (GCS) is also introduced, which efficiently generates a diffuse interface for three-dimensional realistic biomolecules. The performance of the proposed regularization is examined by considering both analytical and GCS diffuse interfaces, and compared with the trilinear method. Moreover, the proposed GCS-regularization algorithm is validated by calculating electrostatic free energies for a set of proteins and by estimating salt affinities for seven protein complexes. The results are consistent with experimental data and estimates of sharp interface PB models.

| [1] |
N. A. Baker, D. Sept, S. Joseph, M. J. Holst, J. A. McCammon, Electrostatics of nanosystems: application to microtubules and the ribosome, Proc. Natl. Acad. Sci., 98 (2001), 10037–10041. doi: 10.1073/pnas.181342398

|
| [2] |
B. Honig, A. Nicholls, Classical electrostatics in biology and chemistry, Science, 268 (1995), 1144–1149. doi: 10.1126/science.7761829
|
| [3] |
A. Nicholls, B. Honig, A rapid finite difference algorithm, utilizing successive over-relaxation to solve the Poisson-Boltzmann equation, J. Comput. Chem., 12 (1991), 435–445. doi: 10.1002/jcc.540120405
|
| [4] | B. Lee, F. M. Richards, Interpretation of protein structure: estimation of static accessibility, J. Molecular Bio., 55 (1973), 379–400. |
| [5] | F. M. Richards, Areas, volumes, packing and protein structure, Ann. Rev. Biophys. Bioeng., 6 (1977), 151–176. |
| [6] |
J. A. Grant, B. Pickup, A Gaussian description of molecular shape, J. Phys. Chem., 99 (1995), 3503–3510. doi: 10.1021/j100011a016
|
| [7] | S. Ahmed-Ullah, S. Zhao, Pseudo-transient ghost fluid methods for the Poisson-Boltzmann equation with a two-component regularization, Appl. Math. Comput., 380 (2020), 125267. |
| [8] |
W. Geng, S. Zhao, A two-component matched interface and boundary (MIB) regularization for charge singularity in implicit solvent, J. Comput. Phys., 351 (2017), 25–39. doi: 10.1016/j.jcp.2017.09.026
|
| [9] |
P. Bates, G.W. Wei, S. Zhao, Minimal molecular surfaces and their applications, J. Comput. Chem., 29 (2008), 380–391. doi: 10.1002/jcc.20796
|
| [10] |
P. Bates, Z. Chen, Y. H. Sun, G. W. Wei, S. Zhao, Geometric and potential driving formation and evolution of biomolecular surfaces, J. Math. Biol., 59 (2009), 193–231. doi: 10.1007/s00285-008-0226-7
|
| [11] |
C. Arghya, Z. Jia, L. Li, S. Zhao, E. Alexov, Reproducing the ensemble average polar solvation energy of a protein from a singlestructure: Gaussian-based smooth dielectric function for macromolecular modeling, J. Chem. Theor. Comput., 14 (2018), 1020–1032. doi: 10.1021/acs.jctc.7b00756
|
| [12] |
T. Hazra, S. Ahmed-Ullah, S. Wang, E. Alexov, S. Zhao, A super-Gaussian Poisson-Boltzmann model for electrostatic solvation free energy calculation: smooth dielectric distribution for protein cavities and in both water and vacuum states, J. Math. Bio., 79 (2019), 631–672. doi: 10.1007/s00285-019-01372-1
|
| [13] |
L. Li, C. Li, Z. Zhang, E. Alexov, On the dielectric "constant" of proteins: smooth dielectric function formacromolecular modeling and its implementation in DelPhi, J. Chem. Theory Comput., 9 (2013), 2126–2136. doi: 10.1021/ct400065j
|
| [14] |
A. Chakravorty, S. Pandey, S. Pahari, S. Zhao, E. Alexov, Capturing the effects of explicit waters in implicit electrostatics modeling: Qualitative justification of Gaussian-based dielectric models in DelPhi, J. Chem. Inf. Modeling, 60 (2020), 2229–2246. doi: 10.1021/acs.jcim.0c00151
|
| [15] |
A. Abrashkin, D. Andelman, H. Orland, Dipolar Poisson-Boltzmann equation: ions and dipoles close to charge interfaces, Phy. Rev. Lett., 99 (2007), 077801. doi: 10.1103/PhysRevLett.99.077801
|
| [16] |
L. T. Cheng, J. Dzubiella, J. A. McCammon, B. Li, Application of the level-set method to the solvation of nonpolar molecules, J. Chem. Phys., 127 (2007), 084503. doi: 10.1063/1.2757169
|
| [17] |
S. Dai, B. Li, J. Liu, Convergence of phase-field free energy and boundary force for molecular solvation, Arch. Ration. Mech. Anal., 227 (2018), 105–147. doi: 10.1007/s00205-017-1158-4
|
| [18] |
Y. Zhao, Y. Y. Kwan, J. Che, B. Li, J. A. McCammon, Phase-field approach to implicit solvation of biomolecules with Coulomb-field approximation, J. Chem. Phys., 139 (2013), 024111. doi: 10.1063/1.4812839
|
| [19] |
L. Chen, M. J. Holst, J. Xu, The finite element approximation of the nonlinear Poisson-Boltzmann equation, SIAM J. Numer. Anal., 45 (2007), 2298–2320. doi: 10.1137/060675514
|
| [20] |
W. Deng, J. Xu, S. Zhao, On developing stable finite element methods for pseudo-time simulation of biomolecular electrostatics, J. Comput. Appl. Math., 330 (2018), 456–474. doi: 10.1016/j.cam.2017.09.004
|
| [21] |
J. A. Grant, B. Pickup, A. Nicholls, A smooth permittivity function for Poisson-Boltzmann solvation methods, J. Comput. Chem., 22 (2001), 608–640. doi: 10.1002/jcc.1032
|
| [22] | M. J. Schnieders, N. A. Baker, P. Ren, J. W. Ponder, Polarizable atomic multipole solutes in a Poisson-Boltzmann continuum, J. Chem. Phys., 126 (2002), 124114. |
| [23] |
R. Egan, F. Gibou, Geometric discretization of the multidimensional Dirac delta distribution - Application to the Poisson equationwith singular source terms, J. Comput. Phys., 346 (2017), 71–90. doi: 10.1016/j.jcp.2017.06.003
|
| [24] | W. Rocchia, E. Alexov, B. Honig, Extending the applicability of the nonlinear Poisson-Boltzmann equation: Multiple dielectric constants and multivalent ions, J. Phys. Chem. B, 105 (2001), 6507–6514. |
| [25] | W. Rocchia, S. Sridharan, A. Nicholls, E. Alexov, A. Chiabrera, B. Honig, Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects, J. Comput. Chem., 23 (2002), 128–137. |
| [26] | P. Benner, V. Khoromskaia, B. Khoromskij, C. Kweyu, M. Stein, Computing electrostatic potentials using regularization based on the range-separated tensor format, preprint, arXiv: 1901.09864v1. |
| [27] |
Q. Cai, J. Wang, H. K. Zhao, R. Luo, On removal of charge singularity in Poisson-Boltzmann equation, J. Chem. Phys., 130 (2009), 145101. doi: 10.1063/1.3099708
|
| [28] | I. L. Chern, J. G. Liu, W. C. Wang, Accurate evaluation of electrostatics for macromolecules in solution, Methods Appl. Anal., 10 (2003), 309–328. |
| [29] |
W. Geng, S. Yu, G. W. Wei, Treatment of charge singularities in implicit solvent models, J. Chem. Phys., 127 (2007), 114106. doi: 10.1063/1.2768064
|
| [30] |
M. Holst, J. A. McCammon, Z. Yu, Y. C. Zhou, Y. Zhu, Adaptive finite element modeling techniques for the Poisson-Boltzmann equation I, Commun. Comput. Phys., 11 (2012), 179–214. doi: 10.4208/cicp.081009.130611a
|
| [31] |
B. Khoromskij, Range-separated tensor decomposition of the discretized Dirac delta and elliptic operator inverse, J. Comput. Phys., 401 (2020), 108998. doi: 10.1016/j.jcp.2019.108998
|
| [32] |
D. Xie, New solution decomposition and minimization schemes for Poisson-Boltzmann equation in calculation of biomolecular electrostatics, J. Comput. Phys., 275 (2014), 294–309. doi: 10.1016/j.jcp.2014.07.012
|
| [33] | Z. Zhou, P. Payne, M. Vasquez, N. Kuhn, M. Levitt, Finite-difference solution of the Poisson-Boltzmann equation: complete elimination of self-energy, J. Comput. Chem., 11 (1996), 1344–1351. |
| [34] | A. Lee, W. Geng, S. Zhao, Regularization methods for the Poisson-Boltzmann equation: comparison and accuracy recovery, J. Comput. Phys., in press, 2021. |
| [35] |
C. Xue, S. Deng, Unified construction of Green's functions for Poisson's equation in inhomogeneous media with diffuse interfaces, J. Comput. Appl. Math., 326 (2017), 296–319. doi: 10.1016/j.cam.2017.06.007
|
| [36] |
S. Wang, A. Lee, E. Alexov, S. Zhao, A regularization approach for solving Poisson's equation with singular charge sources and diffuse interfaces, Appl. Math. Lett., 102 (2020), 106144. doi: 10.1016/j.aml.2019.106144
|
| [37] | M. Holst, The Poisson–Boltzmann Equation: Analysis and Multilevel Numerical Solution, Ph.D thesis, UIUC, 1994. |
| [38] | W. H. Press, S. A. Teukolsky, W. T. Vetterling, B. P. Flannery, Numerical Recipes in Fortran: The Art of Scientific Computing, 2nd edition, Cambridge University Press, 1992. |
| [39] | A. Nicholls, D. L. Mobley, P. J. Guthrie, J. D. Chodera, V. S. Pande, Predicting small-molecule solvation free energies: An informal blind test for computational chemistry, J. Med. Chem. 51 (2008), 769–-779. |
| [40] |
S. Zhao, Operator splitting ADI schemes for pseudo-time coupled nonlinear solvation simulations, J. Comput. Phys., 257 (2014), 1000–1021. doi: 10.1016/j.jcp.2013.09.043
|
| [41] |
C. Bertonati, B. Honig, E. Alexov, Poisson-Boltzmann calculations of nonspecific salt effects on protein-protein binding free energies, Biophy. J., 92 (2007), 1891–1899. doi: 10.1529/biophysj.106.092122
|
| [42] | J. Zha, L. Li, A. Chakravorty, and E. Alexov, Treating ion distribution with Gaussian-based smooth dielectric function in DelPhi, J. Comput. Chem., 38, 1974-1979, (2017). |
Figures(22) / Tables(7)

Siwen Wang, Emil Alexov, Shan Zhao. On regularization of charge singularities in solving the Poisson-Boltzmann equation with a smooth solute-solvent boundary[J]. Mathematical Biosciences and Engineering, 2021, 18(2): 1370-1405. doi: 10.3934/mbe.2021072







 DownLoad:
DownLoad: