Citation: Hatim Machrafi. Nanomedicine by extended non-equilibrium thermodynamics: cell membrane diffusion and scaffold medication release[J]. Mathematical Biosciences and Engineering, 2019, 16(4): 1949-1965. doi: 10.3934/mbe.2019095

| [1] | H. Machrafi, Extended non-equilibrium thermodynamics: from principles to applications in nanosystems, Taylor & Francis Group (CRC Press), (2019). |
| [2] | W. A. Banks, From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery, Nature Rev. Drug Disc., 15 (2016), 275–292. |
| [3] | S. Uppal, K. S. Italiya, D. Chitkara, et al., Nanoparticulate-based drug delivery systems for small molecule anti-diabetic drugs: An emerging paradigm for effective therapy, Acta Biomaterialia, 81 (2018), 20–42. |
| [4] | V. Francia, A. Aliyandi and A. Salvati, Effect of the development of a cell barrier on nanoparticle uptake in endothelial cells, Nanoscale, 10 (2018), 16645–16656. |
| [5] | C. L. Ventola, The nanomedicine revolution: part 1: emerging concepts, Pharm. Therap., 37 (2012), 512–525. |
| [6] | C. H. Lin, C. H. Chen, Z. C. Lin, et al., Recent advances in oral delivery of drugs and bioactive natural products using solid lipid nanoparticles as the carriers, J. Food Drug Analys., 25 (2017), 219–234. |
| [7] | J. Shi, P. W. Kantoff, R. Wooster, et al., Cancer nanomedicine: progress, challenges and opportunities, Nature Rev. Canc., 17 (2017), 20–37. |
| [8] | H. Nehoff, N. N. Parayath, L. Domanovitch, et al., Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect, Int. J. Nanomed., 9 (2014), 2539–2555. |
| [9] | J. Alsenz and E. Haenel, Development of a 7-day, 96-well Caco-2 permeability assay with high-throughput direct UV compound analysis, Pharmac. Res., 20 (2003), 1961–1969. |
| [10] | K. J. Lee, N. Johnson, J. Castelo, et al., Effect of experimental pH on the in vitro permeability in intact rabbit intestines and Caco-2 monolayer, Europ. J. Pharmac. Sci., 25 (2005), 193–200. |
| [11] | V. E. Thiel-Demby, J. E. Humphreys, S. L. A. Williams, et al., Biopharmaceutics classification system: validation and learnings of an in vitro permeability assay, Molec. Pharmac., 6 (2009), 11–18. |
| [12] | K. M. M. Doan, Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs, J. Pharmac. Exp. Therap., 303 (2002), 1029–1037. |
| [13] | P. Garberg, M. Ball, N. Borg, et al., In vitro models for the blood-brain barrier, Toxico. Vitro, 19 (2005), 299–334. |
| [14] | J. M. Diamond and Y. Katz, Interpretation of nonelectrolyte partition coefficients between dimyristoyl lecithin and water, J. Membr. Biol., 17 (1974), 121–154. |
| [15] | K. Bittermann and K. U. Goss, Predicting apparent passive permeability of Caco-2 and MDCK cell-monolayers: A mechanistic model, PLoS ONE, 12 (2017), e0190319. |
| [16] | A. T. Heikkinen, J. Mönkkönen and T. Korjamo, Determination of permeation resistance distribution in in vitro cell monolayer permeation experiments, Europ. J. Pharmac. Sci., 40 (2010), 132–142. |
| [17] | A. Avdeef, Absorption and drug development: solubility, permeability, and charge state, John Wiley & Sons, (2012). |
| [18] | H. Machrafi and G. Lebon, General constitutive equations of heat transport at small length scales and high frequencies with extension to mass and electrical charge transport, Appl. Math. Lett., 52 (2016), 30–37. |
| [19] | D. Jou, J. Casas-Vazquez and G. Lebon, Extended Irreversible Thermodynamics, fourth ed. Springer-Verlag, (2010). |
| [20] | C. Cattaneo, Sulla conduzione del calore, Atti del Sem. Mat. Fis. Univ. Mod., 3 (1948), 83–101. |
| [21] | S. M. Longshaw, M. K. Borg, S. B. Ramisetti, et al., mdFoam+: Advanced molecular dynamics in OpenFOAM, Comp. Phys. Comm., 224 (2018), 1–21. |
| [22] | H. Machrafi and G. Lebon, The role of several heat transfer mechanisms on the enhancement of thermal conductivity in nanofluids, Contin. Mech. Therm., 28 (2016), 1461–1475. |
| [23] | H. Machrafi and G. Lebon, Fluid flow through porous and nanoporous media within the prisme of extended thermodynamics: emphasis on the notion of permeability, Microfluidics Nanofluidics, 22 (2018), 65. |
| [24] | G. Lebon, H. Machrafi, M. Grmela, et al., An extended thermodynamic model of transient heat conduction at sub-continuum scales, Proc. Roy. Soc. A, 467 (2011), 3241–3256. |
| [25] | H. Machrafi, Temperature distribution through a nanofilm by means of a ballistic-diffusive approach, Inventions, 4 (2019), 2. |
| [26] | X. Y. Li, K. Nan, H. Chen, et al., Preparation and characterization of chitosan nanopores membranes for the transport of drugs, Int. J. Pharmac., 420 (2011), 371–377. |
| [27] | S. S. Leung, D. Sindhikara and M. P. Jacobson, Simple predictive models of passive membrane permeability incorporating size-dependent membrane-water partition, J. Chem. Inf. Mod., 56 (2016), 924–929. |
| [28] | R. V. Swift and R.E. Amaro, Modeling the pharmacodynamics of passive membrane permeability, J. Comp.-Aid. Mol. Des., 25 (2011), 1007–1017. |
| [29] | J. Li, C. Fan, H. Pei, et al., Smart drug delivery nanocarriers with self-assembled DNA nanostructures, Adv. Mat., 25 (2013), 4386–4396. |
| [30] | X. Zhang, J. Tu, D. Wang, et al., Programmable and multifunctional DNA-based materials for biomedical applications, Adv. Mat., 30 (2018), 1703658. |
| [31] | X. Qu, S. Wang, Z. Ge, et al., Programming cell adhesion for on-chip sequential boolean logic functions, J. Am. Chem. Soc., 139 (2017), 10176–10179. |
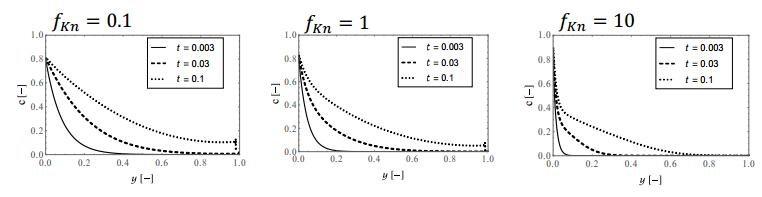
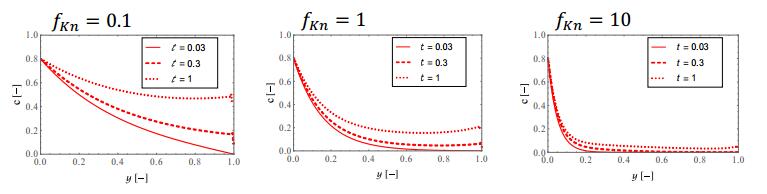
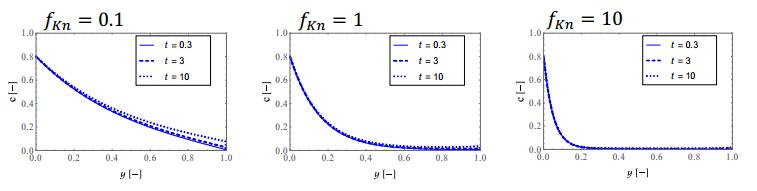
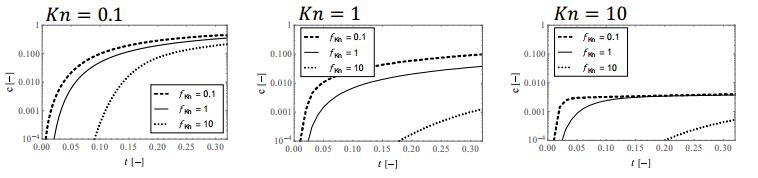
Figures(9)

Hatim Machrafi. Nanomedicine by extended non-equilibrium thermodynamics: cell membrane diffusion and scaffold medication release[J]. Mathematical Biosciences and Engineering, 2019, 16(4): 1949-1965. doi: 10.3934/mbe.2019095







 DownLoad:
DownLoad: