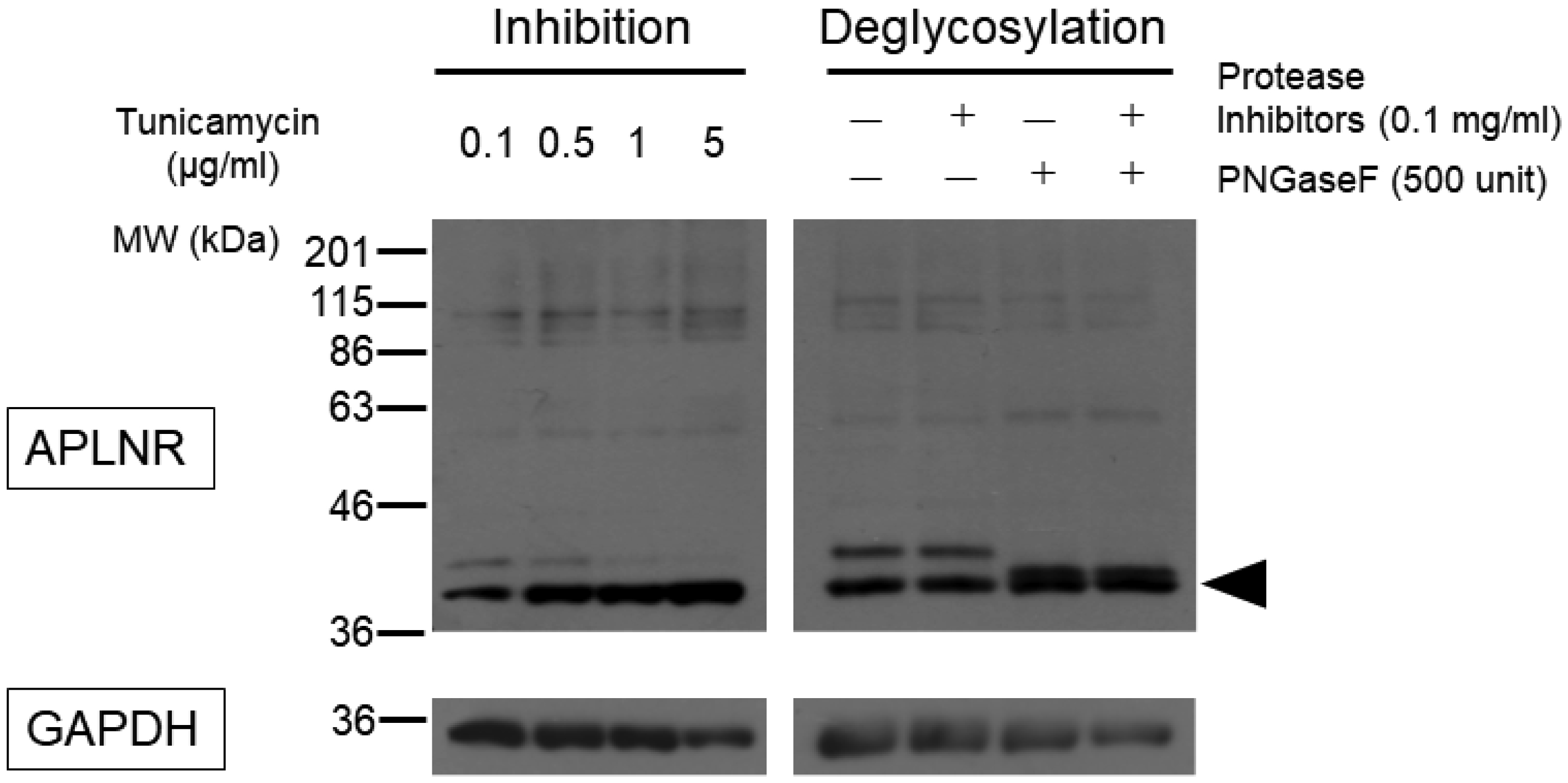
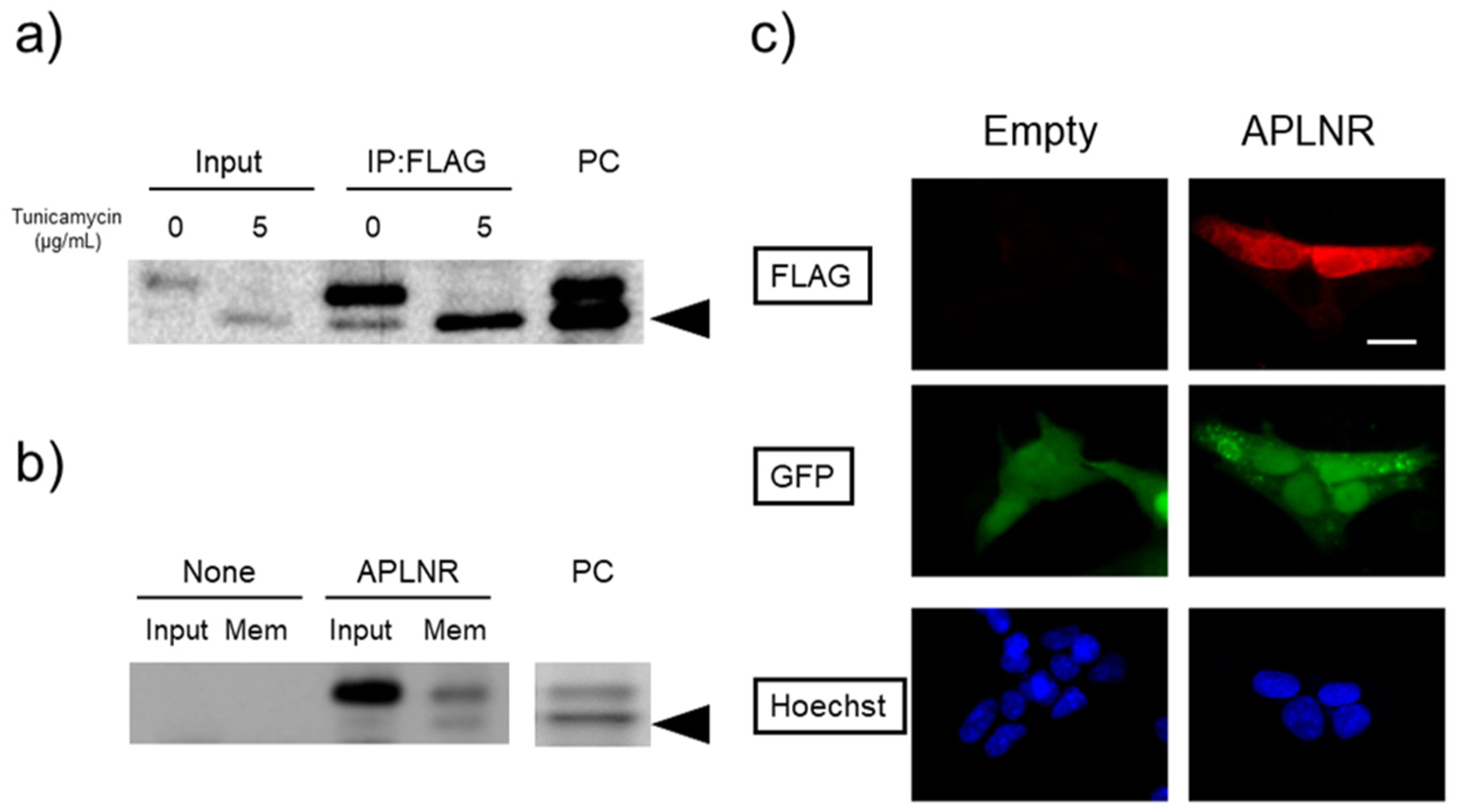
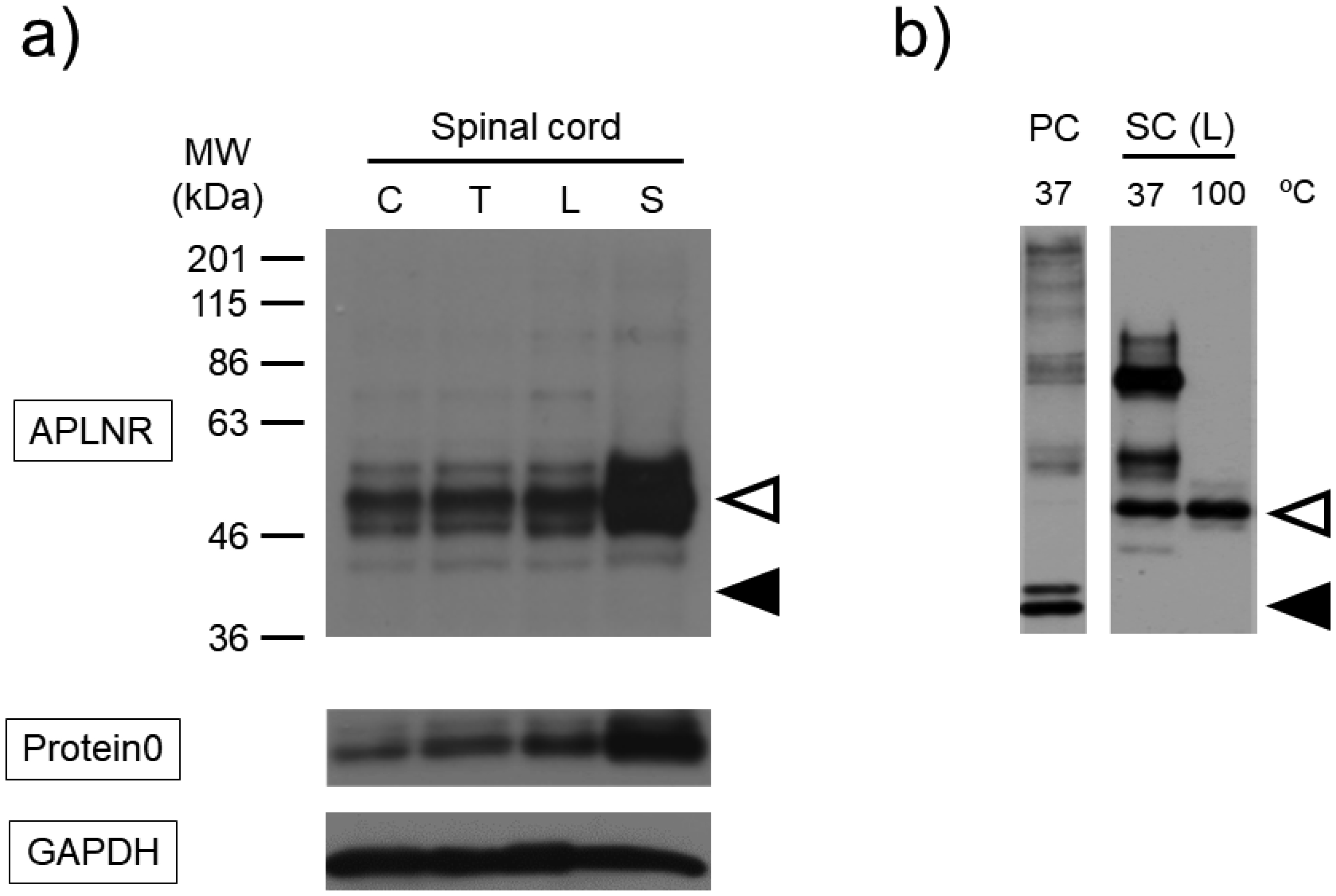
Post-translational modifications (PTMs) are protein modifications that occur after protein biosynthesis, playing a crucial role in regulating protein function. They are involved in the functional expression of G-protein-coupled receptors (GPCRs), as well as intracellular and secretory protein signaling. Here, we aimed to investigate the PTMs of the apelin receptor (APLNR), a GPCR and their potential influence on the receptor's function. In an in vitro experiment using HEK cells, we only observed glycosylation as a PTM of the APLNR and ineffective receptor signaling by the agonist, (Pyr1)-apelin-13. In contrast, when analyzing mouse spinal cord, we detected glycosylation and other PTMs, excluding isopeptidation. This suggests that additional PTMs are involved in the functional expression of the APLNR in vitro. In summary, these findings suggest that the APLNR in vivo requires multiple PTMs for functional expression. To comprehensively understand the pharmacological effects of the APLNR, it is essential to establish an in vitro system that adequately replicates the receptor's PTM profile. Nonetheless, it is crucial to overcome the challenge of heat-sensitive proteolysis in APLNR studies. By elucidating the regulation of PTMs, further research has the potential to advance the analysis and pharmacological studies of both the apelin/APLNR system and GPCR signal modulation.
Citation: Toshihiko Kinjo, Shun Ebisawa, Tatsuya Nokubo, Mifu Hashimoto, Takonori Yamada, Michiko Oshio, Ruka Nakamura, Kyosuke Uno, Nobuyuki Kuramoto. Post-translational modifications of the apelin receptor regulate its functional expression[J]. AIMS Neuroscience, 2023, 10(4): 282-299. doi: 10.3934/Neuroscience.2023022
Post-translational modifications (PTMs) are protein modifications that occur after protein biosynthesis, playing a crucial role in regulating protein function. They are involved in the functional expression of G-protein-coupled receptors (GPCRs), as well as intracellular and secretory protein signaling. Here, we aimed to investigate the PTMs of the apelin receptor (APLNR), a GPCR and their potential influence on the receptor's function. In an in vitro experiment using HEK cells, we only observed glycosylation as a PTM of the APLNR and ineffective receptor signaling by the agonist, (Pyr1)-apelin-13. In contrast, when analyzing mouse spinal cord, we detected glycosylation and other PTMs, excluding isopeptidation. This suggests that additional PTMs are involved in the functional expression of the APLNR in vitro. In summary, these findings suggest that the APLNR in vivo requires multiple PTMs for functional expression. To comprehensively understand the pharmacological effects of the APLNR, it is essential to establish an in vitro system that adequately replicates the receptor's PTM profile. Nonetheless, it is crucial to overcome the challenge of heat-sensitive proteolysis in APLNR studies. By elucidating the regulation of PTMs, further research has the potential to advance the analysis and pharmacological studies of both the apelin/APLNR system and GPCR signal modulation.
protein kinase B
apelin receptor
autophagy-related protein
cyclic adenosine monophosphate
extracellular signal regulated kinase
F locus adjacent transcript 10
forskolin
green Fluorescent Protein
G-protein coupled receptor
glycosylphosphatidylinositol
G protein-coupled receptor 44
human embryonic kidney
interferon-stimulated gene 15
lysophosphatidic acid receptor 1
mitogen-activated protein kinase
mammalian target of rapamycin
neural-precursor-cell-expressed developmentally down-regulated protein
N-methyl-D-aspartate
protease-activated receptor 1
phosphoinositide-dependent protein kinase 1
phosphoinositide 3-kinase
protein kinase A
post-translational modification
sodium dodecyl sulfate polyacrylamide gel electrophoresis
small ubiquitin-like modifier
ubiquitin-fold modifier
ubiquitin-related modifier-1

| [1] |
O'Dowd BF, Heiber M, Chan A, et al. (1993) A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 136: 355-360. https://doi.org/10.1016/0378-1119(93)90495-o 
|
| [2] |
Mughal A, O'Rourke ST (2018) Vascular effects of apelin: Mechanisms and therapeutic potential. Pharmacol Therapeut 190: 139-147. https://doi.org/10.1016/j.pharmthera.2018.05.013
|
| [3] |
Tatemoto K, Hosoya M, Habata Y, et al. (1998) Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Bioph Res Co 251: 471-476. https://doi.org/10.1006/bbrc.1998.9489
|
| [4] |
Jacinto E, Facchinetti V, Liu D, et al. (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127: 125-137. https://doi.org/10.1016/j.cell.2006.08.033
|
| [5] |
Hu J H, Chernoff K, Pelech S, et al. (2003) Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over-expressing mice. J Neurochem 85: 422-431. https://doi.org/10.1046/j.1471-4159.2003.01669.x
|
| [6] |
Ishida J, Hashimoto T, Hashimoto Y, et al. (2004) Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J Biol Chem 279: 26274-26279. https://doi.org/10.1074/jbc.M404149200
|
| [7] |
Kidoya H, Ueno M, Yamada Y, et al. (2008) Spatial and temporal role of the apelin/APJ system in the caliber size regulation of blood vessels during angiogenesis. EMBO J 27: 522-534. https://doi.org/10.1038/sj.emboj.7601982
|
| [8] |
Freyer L, Hsu CW, Nowotschin S, et al. (2017) Loss of Apela peptide in mice causes low penetrance embryonic lethality and defects in early mesodermal derivatives. Cell Rep 20: 2116-2130. https://doi.org/10.1016/j.celrep.2017.08.014
|
| [9] |
Ho L, van Dijk M, Chye STJ, et al. (2017) ELABELA deficiency promotes preeclampsia and cardiovascular malformations in mice. Science (New York, N.Y.) 357: 707-713. https://doi.org/10.1126/science.aam6607
|
| [10] |
Saral S, Topçu A, Alkanat M, et al. (2021) Apelin-13 activates the hippocampal BDNF/TrkB signaling pathway and suppresses neuroinflammation in male rats with cisplatin-induced cognitive dysfunction. Behav Brain Res 408: 113290. https://doi.org/10.1016/j.bbr.2021.113290
|
| [11] |
O'Donnell LA, Agrawal A, Sabnekar P, et al. (2007) Apelin, an endogenous neuronal peptide, protects hippocampal neurons against excitotoxic injury. J Neurochem 102: 1905-1917. https://doi.org/10.1111/j.1471-4159.2007.04645.x
|
| [12] |
Shibagaki F, Ishimaru Y, Sumino A, et al. (2020) Systemic administration of an apelin receptor agonist prevents NMDA-induced loss of retinal neuronal cells in mice. Neurochem Res 45: 752-759. https://doi.org/10.1007/s11064-019-02948-5
|
| [13] |
Kasai A, Kinjo T, Ishihara R, et al. (2011) Apelin deficiency accelerates the progression of amyotrophic lateral sclerosis. PloS One 6: e23968. https://doi.org/10.1371/journal.pone.0023968
|
| [14] |
Pope GR, Tilve S, McArdle CA, et al. (2016) Agonist-induced internalization and desensitization of the apelin receptor. Mol Cell Endocrinol 437: 108-119. https://doi.org/10.1016/j.mce.2016.07.040
|
| [15] | Yang P, Kuc RE, Brame AL, et al. (2017) [Pyr1]Apelin-13(1-12) Is a Biologically Active ACE2 Metabolite of the Endogenous Cardiovascular Peptide [Pyr1]Apelin-13. Front Neurosci 11: 92. https://doi.org/10.3389/fnins.2017.00092 |
| [16] |
de Brevern AG, Rebehmed J (2022) Current status of PTMs structural databases: applications, limitations and prospects. Amino Acids 54: 575-590. https://doi.org/10.1007/s00726-021-03119-z
|
| [17] |
Hay RT (2005) SUMO: A history of modification. Mol Cell 18: 1-12. https://doi.org/10.1016/j.molcel.2005.03.012
|
| [18] |
Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41-45. https://doi.org/10.1038/47412
|
| [19] |
Patwardhan A, Cheng N, Trejo J (2021) Post-translational modifications of G protein-coupled receptors control cellular signaling dynamics in space and time. Pharmacol Rev 73: 120-151. https://doi.org/10.1124/pharmrev.120.000082
|
| [20] |
Kinjo T, Higashi H, Uno K, et al. (2021) Apelin/apelin receptor system: Molecular characteristics, physiological roles, and prospects as a target for disease prevention and pharmacotherapy. Curr Mol Pharmacol 14: 210-219. https://doi.org/10.2174/1874467213666200602133032
|
| [21] |
Kuramoto N, Baba K, Gion K, et al. (2003) Xenobiotic response element binding enriched in both nuclear and microsomal fractions of rat cerebellum. J Neurochem 85: 264-273. https://doi.org/10.1046/j.1471-4159.2003.01679.x
|
| [22] |
Bullis BL, Li X, Singh DN, et al. (2002) Properties of the Na+/H+ exchanger protein. Detergent-resistant aggregation and membrane microdistribution. Eur J Biochem 269: 4887-4895. https://doi.org/10.1046/j.1432-1033.2002.03202.x
|
| [23] |
Kuramoto N, Wilkins ME, Fairfax BP, et al. (2007) Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron 53: 233-247. https://doi.org/10.1016/j.neuron.2006.12.015
|
| [24] |
Puffer BA, Sharron M, Coughlan CM, et al. (2000) Expression and coreceptor function of APJ for primate immunodeficiency viruses. Virology 276: 435-444. https://doi.org/10.1006/viro.2000.0557
|
| [25] |
Ma Y, Yue Y, Ma Y, et al. (2017) Structural basis for apelin control of the human apelin receptor. Structure 25: 858-866.e4. https://doi.org/10.1016/j.str.2017.04.008
|
| [26] |
Wirth M, Schick M, Keller U, et al. (2020) Ubiquitination and ubiquitin-like modifications in multiple myeloma: Biology and therapy. Cancers 12: 3764. https://doi.org/10.3390/cancers12123764
|
| [27] |
Cappadocia L, Lima CD (2018) Ubiquitin-like protein conjugation: Structures, chemistry, and mechanism. Chem Rev 118: 889-918. https://doi.org/10.1021/acs.chemrev.6b00737
|
| [28] |
Paulick MG, Bertozzi CR (2008) The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry 47: 6991-7000. https://doi.org/10.1021/bi8006324
|
| [29] |
Mogha A, D'Rozario M, Monk KR (2016) G protein-coupled receptors in myelinating glia. Trends Pharmacol Sci 37: 977-987. Erratum in:
|
| [30] |
Corell M, Wicher G, Radomska KJ, et al. (2015) GABA and its B-receptor are present at the node of Ranvier in a small population of sensory fibers, implicating a role in myelination. J Neurosci Res 93: 285-295. https://doi.org/10.1002/jnr.23489
|
| [31] |
Shavit E, Beilin O, Korczyn AD, et al. (2008) Thrombin receptor PAR-1 on myelin at the node of Ranvier: a new anatomy and physiology of conduction block. Brain 131: 1113-1122. https://doi.org/10.1093/brain/awn005
|
| [32] |
Martinez-Lopez N, Athonvarangkul D, Mishall P, et al. (2013) Autophagy proteins regulate ERK phosphorylation. Nat Commun 4: 2799. https://doi.org/10.1038/ncomms3799
|
| [33] |
Beck TC, Arhontoulis DC, Morningstar JE, et al. (2022) Cellular and Molecular Mechanisms of MEK1 Inhibitor-Induced Cardiotoxicity. JACC-CardioOncol 4: 535-548. https://doi.org/10.1016/j.jaccao.2022.07.009
|

Figures(8)

Toshihiko Kinjo, Shun Ebisawa, Tatsuya Nokubo, Mifu Hashimoto, Takonori Yamada, Michiko Oshio, Ruka Nakamura, Kyosuke Uno, Nobuyuki Kuramoto. Post-translational modifications of the apelin receptor regulate its functional expression[J]. AIMS Neuroscience, 2023, 10(4): 282-299. doi: 10.3934/Neuroscience.2023022







 DownLoad:
DownLoad: