In the fear memory network, the hippocampus modulates contextual aspects of fear learning while mutual connections between the amygdala and the medial prefrontal cortex are widely involved in fear extinction. G-protein-coupled receptors (GPCRs) are involved in the regulation of fear and anxiety, so the regulation of GPCRs in fear signaling pathways can modulate the mechanisms of fear memory acquisition, consolidation and extinction. Various studies suggested a role of M-type K+ channels in modulating fear expression and extinction, although conflicting data prevented drawing of clear conclusions. In the present work, we examined the impact of M-type K+ channel blockade or activation on contextual fear acquisition and extinction. In addition, regarding the pivotal role of the hippocampus in contextual fear conditioning (CFC) and the involvement of the axon initial segment (AIS) in neuronal plasticity, we investigated whether structural alterations of the AIS in hippocampal neurons occurred during contextual fear memory acquisition and short-time extinction in mice in a behaviorally relevant context.
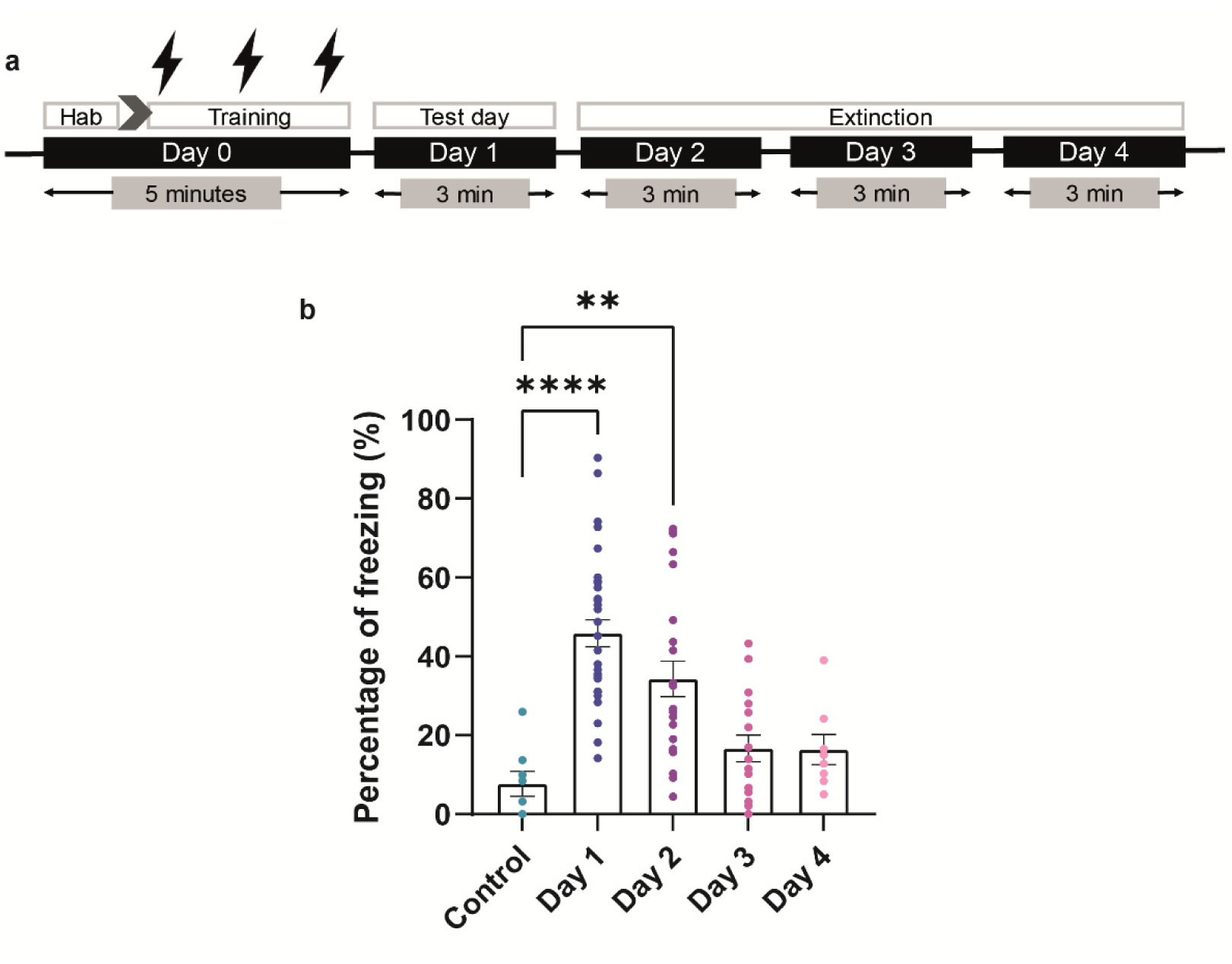
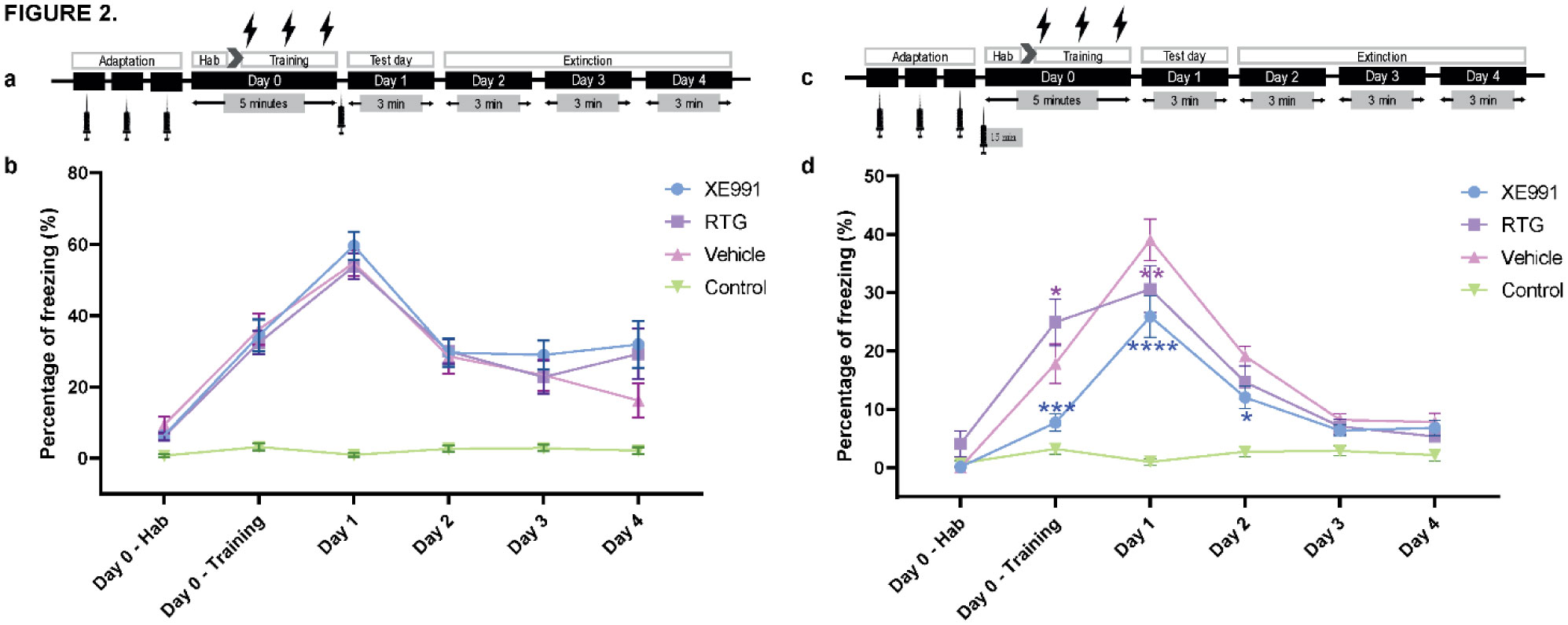
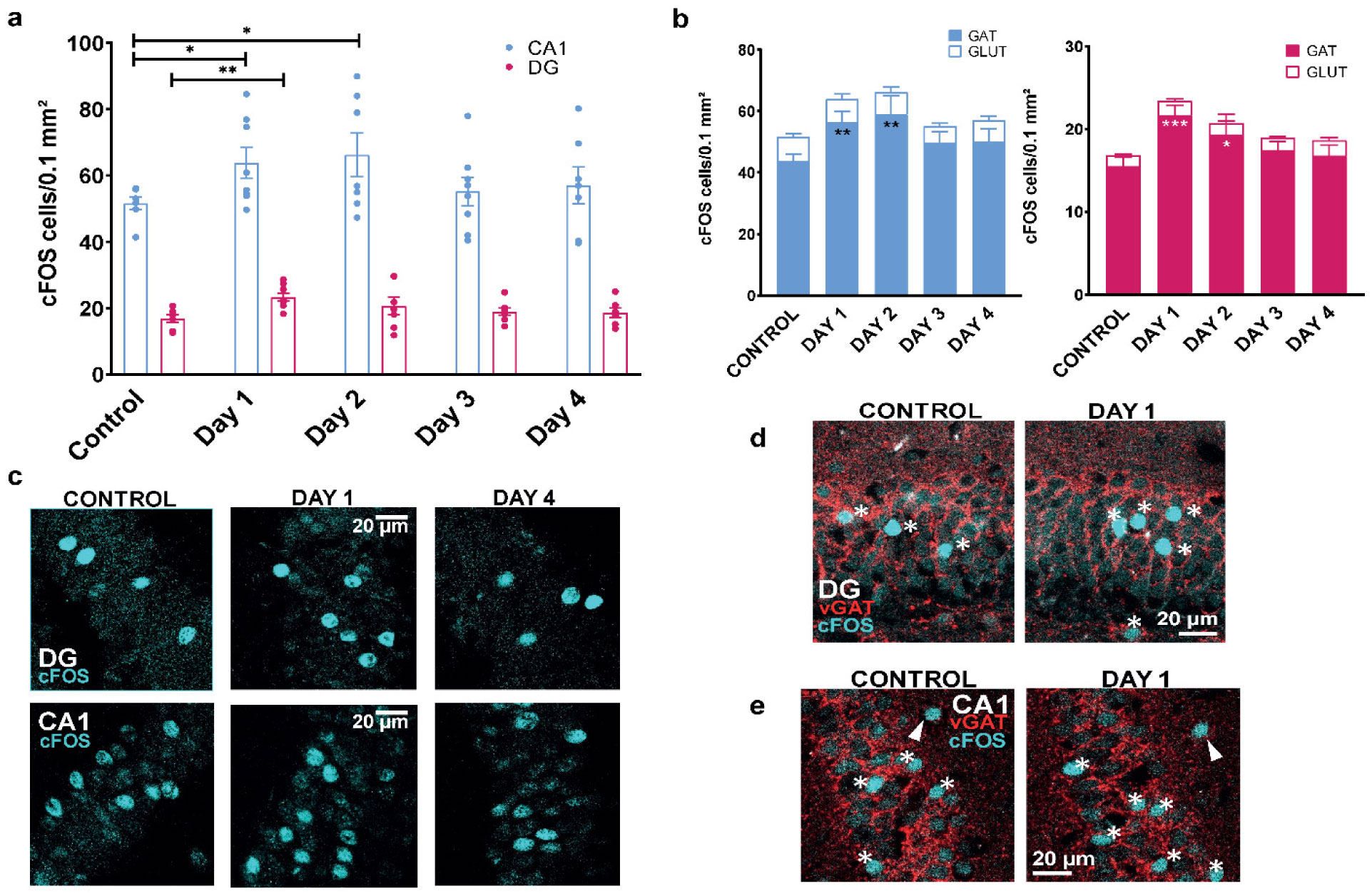
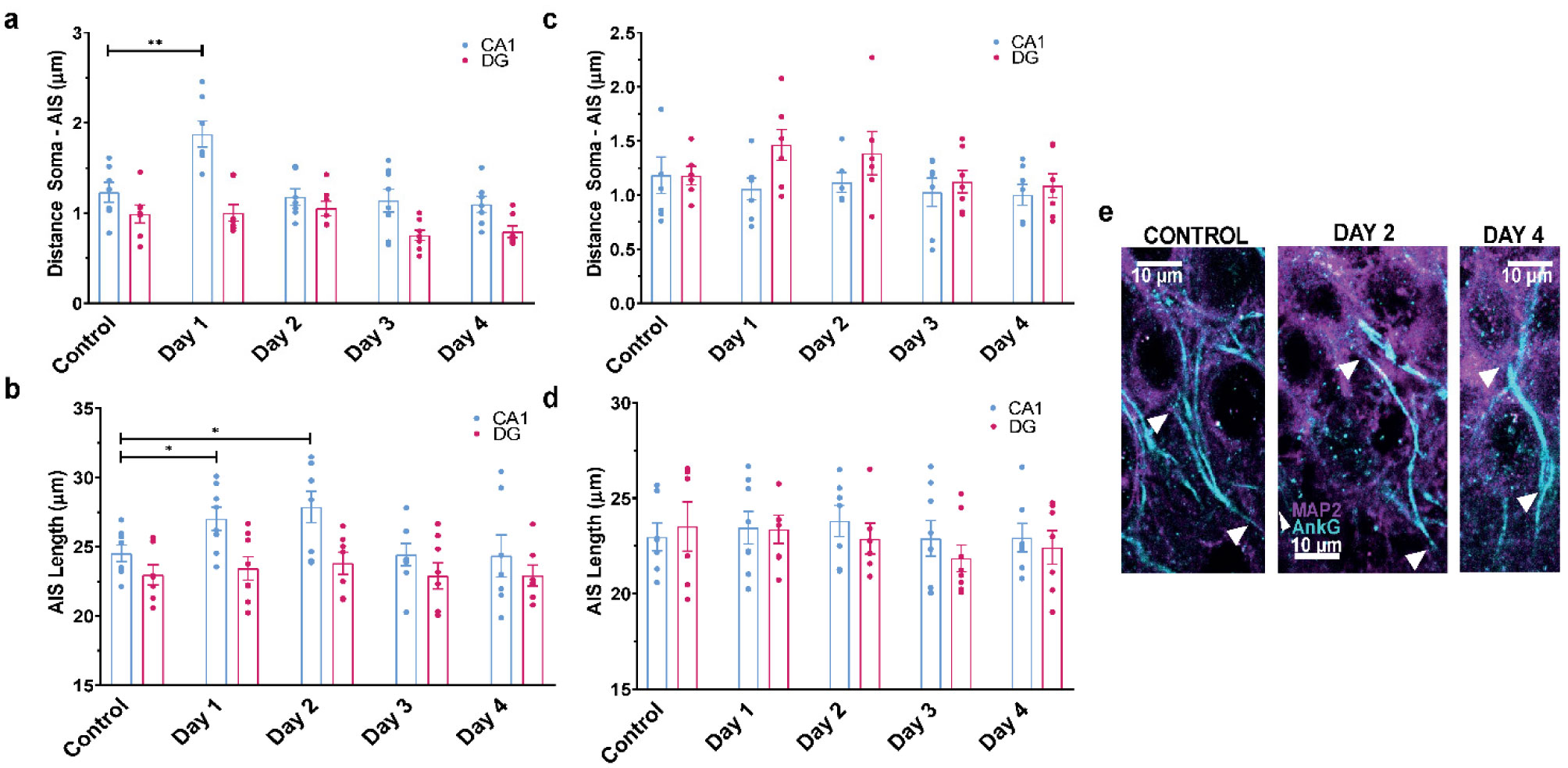
When a single systemic injection of the M-channel blocker XE991 (2 mg/kg, IP) was carried out 15 minutes before the foot shock session, fear expression was significantly reduced. Expression of c-Fos was increased following CFC, mostly in GABAergic neurons at day 1 and day 2 post-fear training in CA1 and dentate gyrus hippocampal regions. A significantly longer AIS segment was observed in GABAergic neurons of the CA1 hippocampal region at day 2.
Our results underscore the role of M-type K + channels in CFC and the importance of hippocampal GABAergic neurons in fear expression.
Citation: Sara Arciniegas Ruiz, Eliav Tikochinsky, Vardit Rubovitch, Chaim G Pick, Bernard Attali. Contextual fear response is modulated by M-type K+ channels and is associated with subtle structural changes of the axon initial segment in hippocampal GABAergic neurons[J]. AIMS Neuroscience, 2023, 10(1): 33-51. doi: 10.3934/Neuroscience.2023003
In the fear memory network, the hippocampus modulates contextual aspects of fear learning while mutual connections between the amygdala and the medial prefrontal cortex are widely involved in fear extinction. G-protein-coupled receptors (GPCRs) are involved in the regulation of fear and anxiety, so the regulation of GPCRs in fear signaling pathways can modulate the mechanisms of fear memory acquisition, consolidation and extinction. Various studies suggested a role of M-type K+ channels in modulating fear expression and extinction, although conflicting data prevented drawing of clear conclusions. In the present work, we examined the impact of M-type K+ channel blockade or activation on contextual fear acquisition and extinction. In addition, regarding the pivotal role of the hippocampus in contextual fear conditioning (CFC) and the involvement of the axon initial segment (AIS) in neuronal plasticity, we investigated whether structural alterations of the AIS in hippocampal neurons occurred during contextual fear memory acquisition and short-time extinction in mice in a behaviorally relevant context.
When a single systemic injection of the M-channel blocker XE991 (2 mg/kg, IP) was carried out 15 minutes before the foot shock session, fear expression was significantly reduced. Expression of c-Fos was increased following CFC, mostly in GABAergic neurons at day 1 and day 2 post-fear training in CA1 and dentate gyrus hippocampal regions. A significantly longer AIS segment was observed in GABAergic neurons of the CA1 hippocampal region at day 2.
Our results underscore the role of M-type K + channels in CFC and the importance of hippocampal GABAergic neurons in fear expression.
Axon initial segment
Ankyrin G
Basolateral amygdala
Contextual fear conditioning
Context Re-exposure Procedure
Conditioned stimulus
Dentate gyrus of hippocampus
G-protein-coupled receptors
Immunohistochemistry
Infralimbic prefrontal cortex
Intraperitoneal
Microtubule-associated protein 2
Medial prefrontal cortex
Retigabine
Unconditioned stimulus
Vesicular GABA transporter
Vesicular glutamate transporter

| [1] | Zelikowsky M, Hersman S, Chawla MK, et al. (2014) Neuronal ensembles in amygdala, hippocampus, and prefrontal cortex track differential components of contextual fear. J Neurosci 34: 8462-8466. https://doi.org/10.1523/JNEUROSCI.3624-13.2014 |
| [2] | Orsini CA, Yan C, Maren S (2013) Ensemble coding of context-dependent fear memory in the amygdala. Front Behav Neurosci 7: 199-199. https://doi.org/10.3389/fnbeh.2013.00199 |
| [3] | Poulos AM, Ponnusamy R, Dong H-W, et al. (2010) Compensation in the neural circuitry of fear conditioning awakens learning circuits in the bed nuclei of the stria terminalis. PNAS 107: 14881-14886. https://doi.org/10.1073/pnas.1005754107 |
| [4] | Helmstetter FJ, Bellgowan PS (1994) Effects of muscimol applied to the basolateral amygdala on acquisition and expression of contextual fear conditioning in rats. Behav Neurosci 108: 1005-1009. https://doi.org/10.1037/0735-7044.108.5.1005 |
| [5] | Muller J, Corodimas KP, Fridel Z, et al. (1997) Functional inactivation of the lateral and basal nuclei of the amygdala by muscimol infusion prevents fear conditioning to an explicit conditioned stimulus and to contextual stimuli. Behav Neurosci 111: 683-691. https://doi.org/10.1037/0735-7044.111.4.683 |
| [6] | Corcoran KA, Quirk GJ (2007) Activity in prelimbic cortex is necessary for the expression of learned, but not innate, fears. J Neurosci 27: 840-844. https://doi.org/10.1523/JNEUROSCI.5327-06.2007 |
| [7] | Goshen I, Brodsky M, Prakash R, et al. (2011) Dynamics of retrieval strategies for remote memories. Cell 147: 678-689. https://doi.org/10.1016/j.cell.2011.09.033 |
| [8] | Zhu LJ, Liu MY, Li H, et al. (2014) The different roles of glucocorticoids in the hippocampus and hypothalamus in chronic stress-induced HPA axis hyperactivity. PLoS One 9: e97689. https://doi.org/10.1371/journal.pone.0097689 |
| [9] | Chaaya N, Battle AR, Johnson LR (2018) An update on contextual fear memory mechanisms: Transition between Amygdala and Hippocampus. Neurosci Biobehav Rev 92: 43-54. https://doi.org/10.1016/j.neubiorev.2018.05.013 |
| [10] | Nees F, Pohlack ST, Grimm O, et al. (2019) White matter correlates of contextual pavlovian fear extinction and the role of anxiety in healthy humans. Cortex 121: 179-188. https://doi.org/10.1016/j.cortex.2019.08.020 |
| [11] | Silva BA, Burns AM, Graff J (2019) A cFos activation map of remote fear memory attenuation. Psychopharmacology (Berl) 236: 369-381. https://doi.org/10.1007/s00213-018-5000-y |
| [12] | Tronson NC, Schrick C, Guzman YF, et al. (2009) Segregated populations of hippocampal principal CA1 neurons mediating conditioning and extinction of contextual fear. J Neurosci 29: 3387-3394. https://doi.org/10.1523/JNEUROSCI.5619-08.2009 |
| [13] | Lebois LAM, Seligowski AV, Wolff JD, et al. (2019) Augmentation of Extinction and Inhibitory Learning in Anxiety and Trauma-Related Disorders. Annu Rev Clin Psychol 15: 257-284. https://doi.org/10.1146/annurev-clinpsy-050718-095634 |
| [14] | Craske MG, Treanor M, Conway CC, et al. (2014) Maximizing exposure therapy: an inhibitory learning approach. Behav Res Ther 58: 10-23. https://doi.org/10.1016/j.brat.2014.04.006 |
| [15] | Myers KM, Ressler KJ, Davis M (2006) Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Memory (Cold Spring Harbor, NY) 13: 216-223. https://doi.org/10.1101/lm.119806 |
| [16] | Westbrook RF, Iordanova M, McNally G, et al. (2002) Reinstatement of fear to an extinguished conditioned stimulus: two roles for context. J Exp Psychol Anim Behav Process 28: 97-110. https://doi.org/10.1037/0097-7403.28.1.97 |
| [17] | Maes JHR, Vossen JMH (1994) The effect of separate reinforced and nonreinforced exposures to a context participating in a Pavlovian discrimination procedure. Elsevier Science, : 231-246. https://doi.org/10.1016/0376-6357(94)90009-4 |
| [18] | Maren S (2001) Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 24: 897-931. https://doi.org/10.1146/annurev.neuro.24.1.897 |
| [19] | Sotres-Bayon F, Quirk GJ (2010) Prefrontal control of fear: more than just extinction. Curr Opin Neurobiol 20: 231-235. https://doi.org/10.1016/j.conb.2010.02.005 |
| [20] | Milad MR, Orr SP, Lasko NB, et al. (2008) Presence and acquired origin of reduced recall for fear extinction in PTSD: results of a twin study. J Psychiatr Res 42: 515-520. https://doi.org/10.1016/j.jpsychires.2008.01.017 |
| [21] | Gowrishankar R, Bruchas MR (2019) Defining circuit-specific roles for G protein-coupled receptors in aversive learning. Curr Opin Behav Sci 26: 146-156. https://doi.org/10.1016/j.cobeha.2019.01.002 |
| [22] | Quirk GJ, Garcia R, Gonzalez-Lima F (2006) Prefrontal mechanisms in extinction of conditioned fear. Biol Psychiatry 60: 337-343. https://doi.org/10.1016/j.biopsych.2006.03.010 |
| [23] | Quirk GJ, Russo GK, Barron JL, et al. (2000) The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 20: 6225-6231. https://doi.org/10.1523/JNEUROSCI.20-16-06225.2000 |
| [24] | Lebron K, Milad MR, Quirk GJ (2004) Delayed recall of fear extinction in rats with lesions of ventral medial prefrontal cortex. Learn Mem 11: 544-548. https://doi.org/10.1101/lm.78604 |
| [25] | Sierra-Mercado D, Corcoran KA, Lebron-Milad K, et al. (2006) Inactivation of the ventromedial prefrontal cortex reduces expression of conditioned fear and impairs subsequent recall of extinction. Eur J Neurosci 24: 1751-1758. https://doi.org/10.1111/j.1460-9568.2006.05014.x |
| [26] | Santini E, Quirk GJ, Porter JT (2008) Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci 28: 4028-4036. https://doi.org/10.1523/JNEUROSCI.2623-07.2008 |
| [27] | Santini E, Porter JT (2010) M-type potassium channels modulate the intrinsic excitability of infralimbic neurons and regulate fear expression and extinction. J Neurosci 30: 12379-12386. https://doi.org/10.1523/JNEUROSCI.1295-10.2010 |
| [28] | Young MB, Thomas SA (2014) M1-muscarinic receptors promote fear memory consolidation via phospholipase C and the M-current. J Neurosci 34: 1570-1578. https://doi.org/10.1523/JNEUROSCI.1040-13.2014 |
| [29] | Fanselow MS (1980) Conditional and Unconditional Components of Post-Shock Freezing. Pavlovian J Biol Sci 15: 177-182. https://doi.org/10.1007/BF03001163 |
| [30] | Anagnostaras SG, Josselyn SA, Frankland PW, et al. (2000) Computer-assisted behavioral assessment of Pavlovian fear conditioning in mice. Learn Mem 7: 58-72. https://doi.org/10.1101/lm.7.1.58 |
| [31] | Cima G (2013) AVMA Guidelines for the Euthanasia of Animal: 2013 Edition. Javma-J Am Vet Med A 242: 715-716. |
| [32] | Alshammari MA, Alshammari TK, Laezza F (2016) Improved Methods for Fluorescence Microscopy Detection of Macromolecules at the Axon Initial Segment. Front Cell Neurosci 10. https://doi.org/10.3389/fncel.2016.00005 |
| [33] | de los Santos-Arteaga M, Sierra-Dominguez SA, Fontanella GH, et al. (2003) Analgesia induced by dietary restriction is mediated by the kappa-opioid system. J Neurosci 23: 11120-11126. https://doi.org/10.1523/JNEUROSCI.23-35-11120.2003 |
| [34] | Lezmy J, Lipinsky M, Khrapunsky Y, et al. (2017) M-current inhibition rapidly induces a unique CK2-dependent plasticity of the axon initial segment. Proc Natl Acad Sci U S A 114: E10234-e10243. https://doi.org/10.1073/pnas.1708700114 |
| [35] | Navarro D, Alvarado M, Navarrete F, et al. (2015) Gestational and early postnatal hypothyroidism alters VGluT1 and VGAT bouton distribution in the neocortex and hippocampus, and behavior in rats. Front Neuroanat 9: 9. https://doi.org/10.3389/fnana.2015.00009 |
| [36] | Kim WB, Cho J-H (2020) Encoding of contextual fear memory in hippocampal–amygdala circuit. Nat Commun 11: 1382. https://doi.org/10.1038/s41467-020-15121-2 |
| [37] | Anagnostaras SG, Gale GD, Fanselow MS (2001) Hippocampus and contextual fear conditioning: Recent controversies and advances. Hippocampus 11: 8-17. https://doi.org/10.1002/1098-1063(2001)11:1<8::AID-HIPO1015>3.0.CO;2-7 |
| [38] | Lara Aparicio SY, Laureani Fierro ÁdJ, Aranda Abreu GE, et al. (2022) Current Opinion on the Use of c-Fos in Neuroscience. NeuroSci 3: 687-702. https://doi.org/10.3390/neurosci3040050 |
| [39] | Milad MR, Quirk GJ (2012) Fear extinction as a model for translational neuroscience: ten years of progress. Ann Rev Psychol 63: 129-151. https://doi.org/10.1146/annurev.psych.121208.131631 |
| [40] | Orsini CA, Maren S (2012) Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav R 36: 1773-1802. https://doi.org/10.1016/j.neubiorev.2011.12.014 |
| [41] | Santini E, Sepulveda-Orengo M, Porter JT (2012) Muscarinic receptors modulate the intrinsic excitability of infralimbic neurons and consolidation of fear extinction. Neuropsychopharmacology 37: 2047-2056. https://doi.org/10.1038/npp.2012.52 |
| [42] | Slomko AM, Naseer Z, Ali SS, et al. (2014) Retigabine calms seizure-induced behavior following status epilepticus. Epilepsy Behav 37: 123-132. https://doi.org/10.1016/j.yebeh.2014.06.010 |
| [43] | Criado-Marrero M, Santini E, Porter JT (2014) Modulating fear extinction memory by manipulating SK potassium channels in the infralimbic cortex. Front Behav Neurosci 8: 96. https://doi.org/10.3389/fnbeh.2014.00096 |
| [44] | Victoria NC, Marron Fernandez de Velasco E, Ostrovskaya O, et al. (2016) G Protein-Gated K(+) Channel Ablation in Forebrain Pyramidal Neurons Selectively Impairs Fear Learning. Biol Psychiatry 80: 796-806. https://doi.org/10.1016/j.biopsych.2015.10.004 |
| [45] | Tischmeyer W, Grimm R (1999) Activation of immediate early genes and memory formation. Cell Mol Life Sci 55: 564-574. https://doi.org/10.1007/s000180050315 |
| [46] | Dragunow M (1996) A role for immediate-early transcription factors in learning and memory. Behav Genet 26: 293-299. https://doi.org/10.1007/BF02359385 |
| [47] | Peng Z, Houser CR (2005) Temporal patterns of fos expression in the dentate gyrus after spontaneous seizures in a mouse model of temporal lobe epilepsy. J Neurosci 25: 7210-7220. https://doi.org/10.1523/JNEUROSCI.0838-05.2005 |
| [48] | Kasugai Y, Vogel E, Hörtnagl H, et al. (2019) Structural and Functional Remodeling of Amygdala GABAergic Synapses in Associative Fear Learning. Neuron 104: 781-794.e784. https://doi.org/10.1016/j.neuron.2019.08.013 |
| [49] | Temel Y, Blokland A, Lim LW (2012) Deactivation of the parvalbumin-positive interneurons in the hippocampus after fear-like behaviour following electrical stimulation of the dorsolateral periaqueductal gray of rats. Behav Brain Res 233: 322-325. https://doi.org/10.1016/j.bbr.2012.05.029 |
| [50] | Caliskan G, Muller I, Semtner M, et al. (2016) Identification of Parvalbumin Interneurons as Cellular Substrate of Fear Memory Persistence. Cereb Cortex 26: 2325-2340. https://doi.org/10.1093/cercor/bhw001 |
| [51] | Whissell PD, Bang JY, Khan I, et al. (2019) Selective Activation of Cholecystokinin-Expressing GABA (CCK-GABA) Neurons Enhances Memory and Cognition. eNeuro 6. https://doi.org/10.1523/ENEURO.0360-18.2019 |
| [52] | Grubb MS, Burrone J (2010) Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465: 1070-1074. https://doi.org/10.1038/nature09160 |
| [53] | Kuba H, Oichi Y, Ohmori H (2010) Presynaptic activity regulates Na+ channel distribution at the axon initial segment. Nature 465: 1075-1078. https://doi.org/10.1038/nature09087 |
| [54] | Grubb MS, Shu Y, Kuba H, et al. (2011) Short- and long-term plasticity at the axon initial segment. J Neurosci 31: 16049-16055. https://doi.org/10.1523/JNEUROSCI.4064-11.2011 |
| [55] | Adachi R, Yamada R, Kuba H (2015) Plasticity of the axonal trigger zone. Neuroscientist 21: 255-265. https://doi.org/10.1177/1073858414535986 |
| [56] | Yamada R, Kuba H (2016) Structural and Functional Plasticity at the Axon Initial Segment. Front Cell Neurosci 10: 250. https://doi.org/10.3389/fncel.2016.00250 |
| [57] | Petersen AV, Cotel F, Perrier JF (2017) Plasticity of the Axon Initial Segment: Fast and Slow Processes with Multiple Functional Roles. Neuroscientist 23: 364-373. https://doi.org/10.1177/1073858416648311 |
| [58] | Leterrier C (2016) Chapter Six - The Axon Initial Segment, 50Years Later: A Nexus for Neuronal Organization and Function. Current Topics in Membranes . Academic Press pp. 185-233. https://doi.org/10.1016/bs.ctm.2015.10.005 |
| [59] | Kole Maarten HP, Stuart Greg J (2012) Signal Processing in the Axon Initial Segment. Neuron 73: 235-247. https://doi.org/10.1016/j.neuron.2012.01.007 |
| [60] | Holmes A, Chen A (2015) GABA receptors in a state of fear. Nat Neurosci 18: 1194-1196. https://doi.org/10.1038/nn.4098 |
| [61] | Rovira-Esteban L, Gunduz-Cinar O, Bukalo O, et al. (2019) Excitation of Diverse Classes of Cholecystokinin Interneurons in the Basal Amygdala Facilitates Fear Extinction. eNeuro 6: ENEURO.0220-0219.2019. https://doi.org/10.1523/ENEURO.0220-19.2019 |
| [62] | Saha R, Knapp S, Chakraborty D, et al. (2017) GABAergic Synapses at the Axon Initial Segment of Basolateral Amygdala Projection Neurons Modulate Fear Extinction. Neuropsychopharmacology 42: 473-484. https://doi.org/10.1038/npp.2016.205 |
Figures(4)

Sara Arciniegas Ruiz, Eliav Tikochinsky, Vardit Rubovitch, Chaim G Pick, Bernard Attali. Contextual fear response is modulated by M-type K+ channels and is associated with subtle structural changes of the axon initial segment in hippocampal GABAergic neurons[J]. AIMS Neuroscience, 2023, 10(1): 33-51. doi: 10.3934/Neuroscience.2023003







 DownLoad:
DownLoad: