Citation: Lin Kooi Ong, Frederick Rohan Walker, Michael Nilsson. Is Stroke a Neurodegenerative Condition? A Critical Review of Secondary Neurodegeneration and Amyloid-beta Accumulation after Stroke[J]. AIMS Medical Science, 2017, 4(1): 1-16. doi: 10.3934/medsci.2017.1.1

| [1] |
Baron JC, Yamauchi H, Fujioka M, et al. (2014) Selective neuronal loss in ischemic stroke and cerebrovascular disease. J Cereb Blood Flow Metab 34: 2-18. doi: 10.1038/jcbfm.2013.188

|
| [2] |
Zhang J, Zhang Y, Xing S, et al. (2012) Secondary neurodegeneration in remote regions after focal cerebral infarction: a new target for stroke management? Stroke 43: 1700-1705. doi: 10.1161/STROKEAHA.111.632448
|
| [3] |
Levine DA, Galecki AT, Langa KM, et al. (2015) Trajectory of Cognitive Decline After Incident Stroke. JAMA 314: 41-51. doi: 10.1001/jama.2015.6968
|
| [4] |
Yang J, Wong A, Wang Z, et al. (2015) Risk factors for incident dementia after stroke and transient ischemic attack. Alzheimers Dement 11: 16-23. doi: 10.1016/j.jalz.2014.01.003
|
| [5] |
Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362: 329-344. doi: 10.1056/NEJMra0909142
|
| [6] |
Caughey B, Lansbury PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26: 267-298. doi: 10.1146/annurev.neuro.26.010302.081142
|
| [7] | Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol 8: 101-112. |
| [8] |
Roychaudhuri R, Yang M, Hoshi MM, et al. (2009) Amyloid beta-protein assembly and Alzheimer disease. J Biol Chem 284: 4749-4753. doi: 10.1074/jbc.R800036200
|
| [9] | Giuffrida ML, Caraci F, De Bona P, et al. (2010) The monomer state of beta-amyloid: where the Alzheimer's disease protein meets physiology. Rev Neurosci 21: 83-93. |
| [10] |
Gilbert BJ (2013) The role of amyloid beta in the pathogenesis of Alzheimer's disease. J Clin Pathol 66: 362-366. doi: 10.1136/jclinpath-2013-201515
|
| [11] | Carrillo-Mora P, Luna R, Colin-Barenque L (2014) Amyloid beta: multiple mechanisms of toxicity and only some protective effects? Oxid Med Cell Longev 2014: 795375. |
| [12] |
Viola KL, Klein WL (2015) Amyloid beta oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathol 129: 183-206. doi: 10.1007/s00401-015-1386-3
|
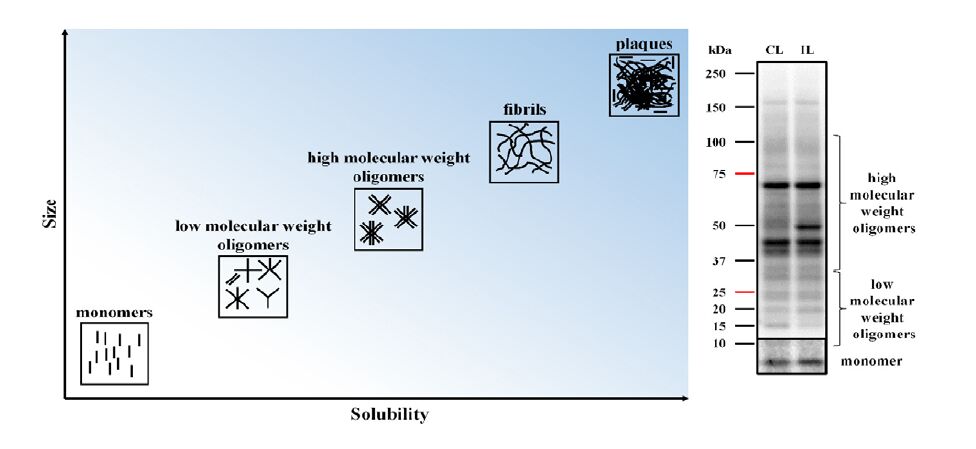
| [13] | Bruggink KA, Muller M, Kuiperij HB, et al. (2012) Methods for analysis of amyloid-beta aggregates. J Alzheimers Dis 28: 735-758. |
| [14] |
Pryor NE, Moss MA, Hestekin CN (2012) Unraveling the early events of amyloid-beta protein (Abeta) aggregation: techniques for the determination of Abeta aggregate size. Int J Mol Sci 13: 3038-3072. doi: 10.3390/ijms13033038
|
| [15] |
van Groen T, Puurunen K, Maki HM, et al. (2005) Transformation of diffuse beta-amyloid precursor protein and beta-amyloid deposits to plaques in the thalamus after transient occlusion of the middle cerebral artery in rats. Stroke 36: 1551-1556. doi: 10.1161/01.STR.0000169933.88903.cf
|
| [16] |
Aho L, Jolkkonen J, Alafuzoff I (2006) Beta-amyloid aggregation in human brains with cerebrovascular lesions. Stroke 37: 2940-2945. doi: 10.1161/01.STR.0000248777.44128.93
|
| [17] |
Makinen S, van Groen T, Clarke J, et al. (2008) Coaccumulation of calcium and beta-amyloid in the thalamus after transient middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab 28: 263-268. doi: 10.1038/sj.jcbfm.9600529
|
| [18] |
Ly JV, Rowe CC, Villemagne VL, et al. (2012) Subacute ischemic stroke is associated with focal 11C PiB positron emission tomography retention but not with global neocortical Abeta deposition. Stroke 43: 1341-1346. doi: 10.1161/STROKEAHA.111.636266
|
| [19] |
Lipsanen A, Kalesnykas G, Pro-Sistiaga P, et al. (2013) Lack of secondary pathology in the thalamus after focal cerebral ischemia in nonhuman primates. Exp Neurol 248: 224-227. doi: 10.1016/j.expneurol.2013.06.016
|
| [20] |
Sahathevan R, Linden T, Villemagne VL, et al. (2016) Positron Emission Tomographic Imaging in Stroke: Cross-Sectional and Follow-Up Assessment of Amyloid in Ischemic Stroke. Stroke 47: 113-119. doi: 10.1161/STROKEAHA.115.010528
|
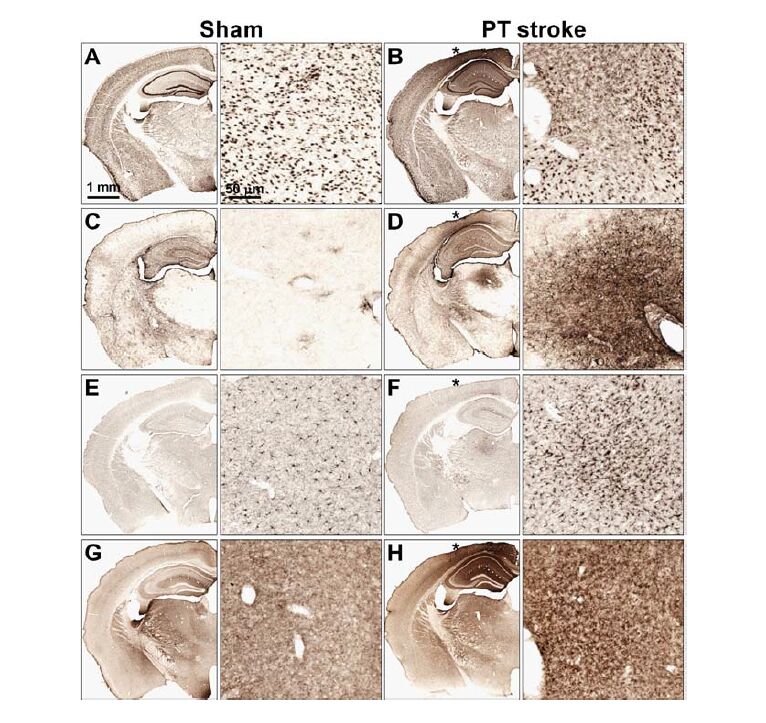
| [21] | Ong LK, Zhao Z, Kluge M, et al. (2016) Chronic stress exposure following photothrombotic stroke is associated with increased levels of Amyloid beta accumulation and altered oligomerisation at sites of thalamic secondary neurodegeneration in mice. J Cereb Blood Flow Metab. |
| [22] |
Lesne S, Koh MT, Kotilinek L, et al. (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440: 352-357. doi: 10.1038/nature04533
|
| [23] |
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8: 595-608. doi: 10.15252/emmm.201606210
|
| [24] | Sheikh S, Safia, Haque E, et al. (2013) Neurodegenerative Diseases: Multifactorial Conformational Diseases and Their Therapeutic Interventions. J Neurodegener Dis 2013: 563481. |
| [25] |
Walsh DM, Klyubin I, Fadeeva JV, et al. (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416: 535-539. doi: 10.1038/416535a
|
| [26] |
Buell AK, Dobson CM, Knowles TP, et al. (2010) Interactions between amyloidophilic dyes and their relevance to studies of amyloid inhibitors. Biophys J 99: 3492-3497. doi: 10.1016/j.bpj.2010.08.074
|
| [27] |
Nesterov EE, Skoch J, Hyman BT, et al. (2005) In vivo optical imaging of amyloid aggregates in brain: design of fluorescent markers. Angew Chem Int Ed Engl 44: 5452-5456. doi: 10.1002/anie.200500845
|
| [28] |
Klunk WE, Engler H, Nordberg A, et al. (2004) Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol 55: 306-319. doi: 10.1002/ana.20009
|
| [29] |
Cohen AD, Rabinovici GD, Mathis CA, et al. (2012) Using Pittsburgh Compound B for in vivo PET imaging of fibrillar amyloid-beta. Adv Pharmacol 64: 27-81. doi: 10.1016/B978-0-12-394816-8.00002-7
|
| [30] |
Liu W, Wong A, Au L, et al. (2015) Influence of Amyloid-beta on Cognitive Decline After Stroke/Transient Ischemic Attack: Three-Year Longitudinal Study. Stroke 46: 3074-3080. doi: 10.1161/STROKEAHA.115.010449
|
| [31] | Rowe CC, Villemagne VL (2011) Brain amyloid imaging. J Nucl Med 52: 1733-1740. |
| [32] |
Glass CK, Saijo K, Winner B, et al. (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140: 918-934. doi: 10.1016/j.cell.2010.02.016
|
| [33] |
Martin JB (1999) Molecular basis of the neurodegenerative disorders. N Engl J Med 340: 1970-1980. doi: 10.1056/NEJM199906243402507
|
| [34] | Kovacs GG (2016) Molecular Pathological Classification of Neurodegenerative Diseases: Turning towards Precision Medicine. Int J Mol Sci 17. |
| [35] |
Tamura A, Tahira Y, Nagashima H, et al. (1991) Thalamic atrophy following cerebral infarction in the territory of the middle cerebral artery. Stroke 22: 615-618. doi: 10.1161/01.STR.22.5.615
|
| [36] |
Ogawa T, Yoshida Y, Okudera T, et al. (1997) Secondary thalamic degeneration after cerebral infarction in the middle cerebral artery distribution: evaluation with MR imaging. Radiology 204: 255-262. doi: 10.1148/radiology.204.1.9205256
|
| [37] |
Nakane M, Tamura A, Sasaki Y, et al. (2002) MRI of secondary changes in the thalamus following a cerebral infarct. Neuroradiology 44: 915-920. doi: 10.1007/s00234-002-0846-3
|
| [38] |
Li C, Ling X, Liu S, et al. (2011) Early detection of secondary damage in ipsilateral thalamus after acute infarction at unilateral corona radiata by diffusion tensor imaging and magnetic resonance spectroscopy. BMC Neurol 11: 49. doi: 10.1186/1471-2377-11-49
|
| [39] |
Buffon F, Molko N, Herve D, et al. (2005) Longitudinal diffusion changes in cerebral hemispheres after MCA infarcts. J Cereb Blood Flow Metab 25: 641-650. doi: 10.1038/sj.jcbfm.9600054
|
| [40] |
Herve D, Molko N, Pappata S, et al. (2005) Longitudinal thalamic diffusion changes after middle cerebral artery infarcts. J Neurol Neurosurg Psychiatry 76: 200-205. doi: 10.1136/jnnp.2004.041012
|
| [41] |
Pappata S, Levasseur M, Gunn RN, et al. (2000) Thalamic microglial activation in ischemic stroke detected in vivo by PET and [11C]PK1195. Neurology 55: 1052-1054. doi: 10.1212/WNL.55.7.1052
|
| [42] |
Gerhard A, Schwarz J, Myers R, et al. (2005) Evolution of microglial activation in patients after ischemic stroke: a [11C](R)-PK11195 PET study. Neuroimage 24: 591-595. doi: 10.1016/j.neuroimage.2004.09.034
|
| [43] |
Thiel A, Heiss WD (2011) Imaging of microglia activation in stroke. Stroke 42: 507-512. doi: 10.1161/STROKEAHA.110.598821
|
| [44] |
Fujie W, Kirino T, Tomukai N, et al. (1990) Progressive shrinkage of the thalamus following middle cerebral artery occlusion in rats. Stroke 21: 1485-1488. doi: 10.1161/01.STR.21.10.1485
|
| [45] |
Iizuka H, Sakatani K, Young W (1990) Neural damage in the rat thalamus after cortical infarcts. Stroke 21: 790-794. doi: 10.1161/01.STR.21.5.790
|
| [46] |
Dihne M, Grommes C, Lutzenburg M, et al. (2002) Different mechanisms of secondary neuronal damage in thalamic nuclei after focal cerebral ischemia in rats. Stroke 33: 3006-3011. doi: 10.1161/01.STR.0000039406.64644.CB
|
| [47] |
Justicia C, Ramos-Cabrer P, Hoehn M (2008) MRI detection of secondary damage after stroke: chronic iron accumulation in the thalamus of the rat brain. Stroke 39: 1541-1547. doi: 10.1161/STROKEAHA.107.503565
|
| [48] |
Bihel E, Pro-Sistiaga P, Letourneur A, et al. (2010) Permanent or transient chronic ischemic stroke in the non-human primate: behavioral, neuroimaging, histological, and immunohistochemical investigations. J Cereb Blood Flow Metab 30: 273-285. doi: 10.1038/jcbfm.2009.209
|
| [49] |
Ross DT, Ebner FF (1990) Thalamic retrograde degeneration following cortical injury: an excitotoxic process? Neuroscience 35: 525-550. doi: 10.1016/0306-4522(90)90327-Z
|
| [50] |
Jones KA, Zouikr I, Patience M, et al. (2015) Chronic stress exacerbates neuronal loss associated with secondary neurodegeneration and suppresses microglial-like cells following focal motor cortex ischemia in the mouse. Brain Behav Immun 48: 57-67. doi: 10.1016/j.bbi.2015.02.014
|
| [51] |
Patience MJ, Zouikr I, Jones K, et al. (2015) Photothrombotic stroke induces persistent ipsilateral and contralateral astrogliosis in key cognitive control nuclei. Neurochem Res 40: 362-371. doi: 10.1007/s11064-014-1487-8
|
| [52] |
Hiltunen M, Makinen P, Peraniemi S, et al. (2009) Focal cerebral ischemia in rats alters APP processing and expression of Abeta peptide degrading enzymes in the thalamus. Neurobiol Dis 35: 103-113. doi: 10.1016/j.nbd.2009.04.009
|
| [53] |
Zhang Y, Xing S, Zhang J, et al. (2011) Reduction of beta-amyloid deposits by gamma-secretase inhibitor is associated with the attenuation of secondary damage in the ipsilateral thalamus and sensory functional improvement after focal cortical infarction in hypertensive rats. J Cereb Blood Flow Metab 31: 572-579. doi: 10.1038/jcbfm.2010.127
|
| [54] |
Sarajarvi T, Lipsanen A, Makinen P, et al. (2012) Bepridil decreases Abeta and calcium levels in the thalamus after middle cerebral artery occlusion in rats. J Cell Mol Med 16: 2754-2767. doi: 10.1111/j.1582-4934.2012.01599.x
|
| [55] |
Zhang J, Zhang Y, Li J, et al. (2012) Autophagosomes accumulation is associated with beta-amyloid deposits and secondary damage in the thalamus after focal cortical infarction in hypertensive rats. J Neurochem 120: 564-573. doi: 10.1111/j.1471-4159.2011.07496.x
|
| [56] |
Mitkari B, Kerkela E, Nystedt J, et al. (2015) Unexpected complication in a rat stroke model: exacerbation of secondary pathology in the thalamus by subacute intraarterial administration of human bone marrow-derived mesenchymal stem cells. J Cereb Blood Flow Metab 35: 363-366. doi: 10.1038/jcbfm.2014.235
|
Figures(2) / Tables(3)

Lin Kooi Ong, Frederick Rohan Walker, Michael Nilsson. Is Stroke a Neurodegenerative Condition? A Critical Review of Secondary Neurodegeneration and Amyloid-beta Accumulation after Stroke[J]. AIMS Medical Science, 2017, 4(1): 1-16. doi: 10.3934/medsci.2017.1.1







 DownLoad:
DownLoad: