Citation: Mayuko Ichimura, Akari Minami, Noriko Nakano, Yasuko Kitagishi, Toshiyuki Murai, Satoru Matsuda. Cigarette smoke may be an exacerbation factor in nonalcoholic fatty liver disease via modulation of the PI3K/AKT pathway[J]. AIMS Molecular Science, 2015, 2(4): 427-439. doi: 10.3934/molsci.2015.4.427

| [1] |
Fargion S, Porzio M, Fracanzani AL (2014) Nonalcoholic fatty liver disease and vascular disease: state-of-the-art. World J Gastroenterol 20: 13306-13324. doi: 10.3748/wjg.v20.i37.13306

|
| [2] | Hjelkrem MC, Torres DM, Harrison SA (2008) Nonalcoholic fatty liver disease. Minerva Med 99: 583-593. |
| [3] | Brandão DF, Ramalho LN, Ramalho FS, et al. (2006) Liver cirrhosis and hepatic stellate cells. Acta Cir Bras 21 Suppl 1: 54-57. |
| [4] |
Nseir W, Shalata A, Marmor A, et al. (2011) Mechanisms linking nonalcoholic fatty liver disease with coronary artery disease. Dig Dis Sci 56: 3439-3449. doi: 10.1007/s10620-011-1767-y
|
| [5] |
Strange RC, Shipman KE, Ramachandran S (2015) Metabolic syndrome: A review of the role of vitamin D in mediating susceptibility and outcome. World J Diabetes 6: 896-911. doi: 10.4239/wjd.v6.i7.896
|
| [6] | Ganji SH, Kashyap ML, Kamanna VS (2015) Niacin inhibits fat accumulation, oxidative stress, and inflammatory cytokine IL-8 in cultured hepatocytes: Impact on non-alcoholic fatty liver disease. Metabolism pii: S0026-0495(15)00134-1. |
| [7] |
Mallat A, Lotersztajn S (2009) Cigarette smoke exposure: a novel cofactor of NAFLD progression? J Hepatol 51: 430-432. doi: 10.1016/j.jhep.2009.05.021
|
| [8] |
Friis-Liby I, Aldenborg F, Jerlstad P, et al. (2004) High prevalence of metabolic complications in patients with non-alcoholic fatty liver disease. Scand J Gastroenterol 39: 864-869. doi: 10.1080/00365520410006431
|
| [9] |
Karim ZA, Alshbool FZ, Vemana HP, et al. (2015) Third-hand Smoke: Impact on Hemostasis and Thrombogenesis. J Cardiovasc Pharmacol 66: 177-182. doi: 10.1097/FJC.0000000000000260
|
| [10] | Chavez-Tapia NC, Barrientos-Gutierrez T, Tellez-Avila FI, et al. (2006) Insulin sensitizers in treatment of nonalcoholic fatty liver disease: Systematic review. World J Gastroenterol 12: 7826-7831. |
| [11] |
Athyros VG, Tziomalos K, Katsiki N, et al. (2013) The impact of smoking on cardiovascular outcomes and comorbidities in statin-treated patients with coronary artery disease: a post hoc analysis of the GREACE study. Curr Vasc Pharmacol 11: 779-784. doi: 10.2174/1570161111311050016
|
| [12] |
Liu Y, Dai M, Bi Y, et al. (2013) Active smoking, passive smoking, and risk of nonalcoholic fatty liver disease (NAFLD): a population-based study in China. J Epidemiol 23: 115-121. doi: 10.2188/jea.JE20120067
|
| [13] | Oniki K, Hori M, Saruwatari J, et al. (2013) Interactive effects of smoking and glutathione S-transferase polymorphisms on the development of non-alcoholic fatty liver disease. Toxicol Lett 220: 143-149. |
| [14] |
Stein LL, Dong MH, Loomba R (2009) Insulin sensitizers in nonalcoholic fatty liver disease and steatohepatitis: Current status. Adv Ther 26: 893-907. doi: 10.1007/s12325-009-0072-z
|
| [15] | Tokuhira N, Kitagishi Y, Suzuki M, et al. (2015) PI3K/AKT/PTEN pathway as a target for Crohn's disease therapy (Review). Int J Mol Med 35: 10-16. |
| [16] | Duan J, Yu Y, Yu Y, et al. (2014) Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/mTOR signaling pathway. Int J Nanomedicine 9: 5131-5141. |
| [17] |
Song BQ, Chi Y, Li X, et al. (2015) Inhibition of Notch Signaling Promotes the Adipogenic Differentiation of Mesenchymal Stem Cells Through Autophagy Activation and PTEN-PI3K/AKT/mTOR Pathway. Cell Physiol Biochem 36: 1991-2002. doi: 10.1159/000430167
|
| [18] |
Wang Z, Li N, Wang B, et al. (2015) Nonalcoholic fatty liver disease progression in rats is accelerated by splenic regulation of liver PTEN/AKT. Saudi J Gastroenterol 21: 232-238. doi: 10.4103/1319-3767.161641
|
| [19] |
Ziamajidi N, Khaghani S, Hassanzadeh G, et al. (2013) Amelioration by chicory seed extract of diabetes- and oleic acid-induced non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH) via modulation of PPARα and SREBP-1. Food Chem Toxicol 58: 198-209. doi: 10.1016/j.fct.2013.04.018
|
| [20] |
Serviddio G, Sastre J, Bellanti F, et al. (2008) Mitochondrial involvement in non-alcoholic steatohepatitis. Mol Aspects Med 29: 22-35. doi: 10.1016/j.mam.2007.09.014
|
| [21] |
Roy D, Cai Q, Felty Q, et al. (2007) Estrogen-induced generation of reactive oxygen and nitrogen species, gene damage, and estrogen-dependent cancers. J Toxicol Environ Health B Crit Rev 10: 235-257. doi: 10.1080/15287390600974924
|
| [22] |
Futosi K, Fodor S, Mócsai A (2013) Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol 17: 638-650. doi: 10.1016/j.intimp.2013.06.034
|
| [23] |
Masamune A, Shimosegawa T (2009) Signal transduction in pancreatic stellate cells. J Gastroenterol 44: 249-260. doi: 10.1007/s00535-009-0013-2
|
| [24] |
Yao H, Han X, Han X (2014) The cardioprotection of the insulin-mediated PI3K/Akt/mTOR signaling pathway. Am J Cardiovasc Drugs 14: 433-442. doi: 10.1007/s40256-014-0089-9
|
| [25] | Chen Z, Wang D, Liu X, et al. (2015) Oxidative DNA damage is involved in cigarette smoke-induced lung injury in rats. Environ Health Prev Med [Epub ahead of print] |
| [26] |
Azzalini L, Ferrer E, Ramalho LN, et al. (2010) Cigarette smoking exacerbates nonalcoholic fatty liver disease in obese rats. Hepatology 51: 1567-1576. doi: 10.1002/hep.23516
|
| [27] |
Balti EV, Echouffo-Tcheugui JB, Yako YY, et al. (2014) Air pollution and risk of type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetes Res Clin Pract 106: 161-172. doi: 10.1016/j.diabres.2014.08.010
|
| [28] |
Wei X, E M, Yu S (2015) A meta-analysis of passive smoking and risk of developing Type 2 Diabetes Mellitus. Diabetes Res Clin Pract 107: 9-14. doi: 10.1016/j.diabres.2014.09.019
|
| [29] | Bhatt HB, Smith RJ (2015) Fatty liver disease in diabetes mellitus. Hepatobiliary Surg Nutr 4: 101-108. |
| [30] |
Collin A, Hardonnière K, Chevanne M, et al. (2014) Cooperative interaction of benzo[a]pyrene and ethanol on plasma membrane remodeling is responsible for enhanced oxidative stress and cell death in primary rat hepatocytes. Free Radic Biol Med 72: 11-22. doi: 10.1016/j.freeradbiomed.2014.03.029
|
| [31] |
Chen H, Hansen MJ, Jones JE, et al. (2007) Detrimental metabolic effects of combining long-term cigarette smoke exposure and high-fat diet in mice. Am J Physiol Endocrinol Metab 293: E1564-E1571. doi: 10.1152/ajpendo.00442.2007
|
| [32] |
Drehmer JE, Ossip DJ, Rigotti NA, et al. (2012) Pediatrician interventions and thirdhand smoke beliefs of parents. Am J Prev Med 43: 533-536. doi: 10.1016/j.amepre.2012.07.020
|
| [33] |
Sleiman M, Gundel LA, Pankow JF, et al. (2010) Formation of carcinogens indoors by surface-mediated reactions of nicotine with nitrous acid, leading to potential thirdhand smoke hazards. Proc Natl Acad Sci U S A 107: 6576-6581. doi: 10.1073/pnas.0912820107
|
| [34] |
Mizoue T, Ueda R, Hino Y, et al. (1999) Workplace exposure to environmental tobacco smoke and high density lipoprotein cholesterol among nonsmokers. Am J Epidemiol 150: 1068-1072. doi: 10.1093/oxfordjournals.aje.a009930
|
| [35] |
Tannapfel A, Denk H, Dienes HP, et al. (2011) Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Virchows Arch 458: 511-523. doi: 10.1007/s00428-011-1066-1
|
| [36] |
Brait M, Munari E, LeBron C, et al. (2013) Genome-wide methylation profiling and the PI3K-AKT pathway analysis associated with smoking in urothelial cell carcinoma. Cell Cycle 12: 1058-1070. doi: 10.4161/cc.24050
|
| [37] |
Ibuki Y, Toyooka T, Zhao X, et al. (2014) Cigarette sidestream smoke induces histone H3 phosphorylation via JNK and PI3K/Akt pathways, leading to the expression of proto-oncogenes. Carcinogenesis 35: 1228-1237. doi: 10.1093/carcin/bgt492
|
| [38] |
Park CH, Lee IS, Grippo P, et al. (2013) Akt kinase mediates the prosurvival effect of smoking compounds in pancreatic ductal cells. Pancreas 42: 655-662. doi: 10.1097/MPA.0b013e3182762928
|
| [39] |
Hosgood HD 3rd, Menashe I, He X, et al. (2009) PTEN identified as important risk factor of chronic obstructive pulmonary disease. Respir Med 103: 1866-1870. doi: 10.1016/j.rmed.2009.06.016
|
| [40] |
Hoffmann RF, Zarrintan S, Brandenburg SM, et al. (2013) Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res 14: 97. doi: 10.1186/1465-9921-14-97
|
| [41] |
Zhou X, Zhao L, Mao J, et al. (2015) Antioxidant effects of hydrogen sulfide on left ventricular remodeling in smoking rats are mediated via PI3K/Akt-dependent activation of Nrf2. Toxicol Sci 144: 197-203. doi: 10.1093/toxsci/kfu272
|
| [42] |
Aravamudan B, Kiel A, Freeman M, et al. (2014) Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 306: L840-L854. doi: 10.1152/ajplung.00155.2013
|
| [43] |
Rutherford C, Childs S, Ohotski J, et al. (2013) Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis 4: e927. doi: 10.1038/cddis.2013.455
|
| [44] |
Hawkins PT, Anderson KE, Davidson K, et al. (2006) Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans 34: 647-662. doi: 10.1042/BST0340647
|
| [45] |
Murugan AK, Munirajan AK, Tsuchida N (2013) Genetic deregulation of the PIK3CA oncogene in oral cancer. Cancer Lett 338: 193-203. doi: 10.1016/j.canlet.2013.04.005
|
| [46] |
Dong H, Zhang X, Dai X, et al. (2014) Lithium ameliorates lipopolysaccharide-induced microglial activation via inhibition of toll-like receptor 4 expression by activating the PI3K/Akt/FoxO1 pathway. J Neuroinflammation 11: 140. doi: 10.1186/s12974-014-0140-4
|
| [47] |
Peres AG, Stegen C, Li J, et al. (2015) Uncoupling of pro- and anti-inflammatory properties of Staphylococcus aureus. Infect Immun 83: 1587-1597. doi: 10.1128/IAI.02832-14
|
| [48] |
Edirisinghe I, Rahman I (2010) Cigarette smoke-mediated oxidative stress, shear stress, and endothelial dysfunction: role of VEGFR2. Ann N Y Acad Sci 1203: 66-72. doi: 10.1111/j.1749-6632.2010.05601.x
|
| [49] | Ahluwalia A, Tarnawski AS (2012) Critical role of hypoxia sensor--HIF-1α in VEGF gene activation. Implications for angiogenesis and tissue injury healing. Curr Med Chem 19: 90-97. |
| [50] | Tarantino G, Lobello R, Scopacasa F, et al. (2007) The contribution of omental adipose tissue to adipokine concentrations in patients with the metabolic syndrome. Clin Invest Med 30: E192-E199. |
| [51] |
Tarantino G, Conca P, Pasanisi F, et al. (2009) Could inflammatory markers help diagnose nonalcoholic steatohepatitis? Eur J Gastroenterol Hepatol 21: 504-511. doi: 10.1097/MEG.0b013e3283229b40
|
| [52] |
Tarantino G, Capone D (2013) Inhibition of the mTOR pathway: a possible protective role in coronary artery disease. Ann Med 45: 348-356. doi: 10.3109/07853890.2013.770333
|
| [53] |
Cozzone D, Fröjdö S, Disse E, et al. (2008) Isoform-specific defects of insulin stimulation of Akt/protein kinase B (PKB) in skeletal muscle cells from type 2 diabetic patients. Diabetologia 51: 512-521. doi: 10.1007/s00125-007-0913-8
|
| [54] |
Rane MJ, Coxon PY, Powell DW, et al. (2001) p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J Biol Chem 276: 3517-3523. doi: 10.1074/jbc.M005953200
|
| [55] |
Knobbe CB, Merlo A, Reifenberger G (2002) Pten signaling in gliomas. Neuro Oncol 4: 196-211. doi: 10.1215/15228517-4-3-196
|
| [56] | Georgescu MM, Kirsch KH, Kaloudis P, et al. (2000) Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res 60: 7033-7038. |
| [57] | Yoshida H, Okumura N, Kitagishi Y, et al. (2011) Ethanol extract of Rosemary repressed PTEN expression in K562 culture cells. Int J appl Biol pharm Technol 2: 316-322. |
| [58] |
Tamguney T, Stokoe D (2007) New insights into PTEN. J Cell Sci 120: 4071-4079. doi: 10.1242/jcs.015230
|
| [59] |
Sato W, Horie Y, Kataoka E, et al. (2006) Hepatic gene expression in hepatocyte-specific Pten deficient mice showing steatohepatitis without ethanol challenge. Hepatol Res 34: 256-265. doi: 10.1016/j.hepres.2006.01.003
|
| [60] | Piguet AC, Stroka D, Zimmermann A, et al. (2009) Hypoxia aggravates non-alcoholic steatohepatitis in mice lacking hepatocellular PTEN. Clin Sci (Lond) 118: 401-410. |
| [61] |
Li Y, Hai J, Li L, et al. (2013) Administration of ghrelin improves inflammation, oxidative stress, and apoptosis during and after non-alcoholic fatty liver disease development. Endocrine 43: 376-386. doi: 10.1007/s12020-012-9761-5
|
| [62] |
Ikejima K, Okumura K, Lang T, et al. (2005) The role of leptin in progression of non-alcoholic fatty liver disease. Hepatol Res 33: 151-154. doi: 10.1016/j.hepres.2005.09.024
|
| [63] |
Suman S, Kallakury BV, Fornace AJ Jr, et al. (2015) Protracted upregulation of leptin and IGF1 is associated with activation of PI3K/Akt and JAK2 pathway in mouse intestine after ionizing radiation exposure. Int J Biol Sci 11: 274-283. doi: 10.7150/ijbs.10684
|
| [64] | Imajo K, Fujita K, Yoneda M, et al. (2012) Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab16:44-54. |
| [65] |
Waseem T, Duxbury M, Ashley SW, et al. (2014) Ghrelin promotes intestinal epithelial cell proliferation through PI3K/Akt pathway and EGFR trans-activation both converging to ERK 1/2 phosphorylation. Peptides 52: 113-121. doi: 10.1016/j.peptides.2013.11.021
|
| [66] |
Tomoda K, Kubo K, Nishii Y, et al. (2012) Changes of ghrelin and leptin levels in plasma by cigarette smoke in rats. J Toxicol Sci 37: 131-138. doi: 10.2131/jts.37.131
|
| [67] |
Zhang F, Zhang Z, Kong D, et al. (2014) Tetramethylpyrazine reduces glucose and insulin-induced activation of hepatic stellate cells by inhibiting insulin receptor-mediated PI3K/AKT and ERK pathways. Mol Cell Endocrinol 382: 197-204. doi: 10.1016/j.mce.2013.09.020
|
| [68] |
Dal-Cim T, Molz S, Egea J, et al. (2012) Guanosine protects human neuroblastoma SH-SY5Y cells against mitochondrial oxidative stress by inducing heme oxigenase-1 via PI3K/Akt/GSK-3β pathway. Neurochem Int 61: 397-404. doi: 10.1016/j.neuint.2012.05.021
|
| [69] |
Kudo Y, Tanaka Y, Tateishi K, et al. (2011) Altered composition of fatty acids exacerbates hepatotumorigenesis during activation of the phosphatidylinositol 3-kinase pathway. J Hepatol 55: 1400-1408. doi: 10.1016/j.jhep.2011.03.025
|
| [70] |
Santos PP, Oliveira F, Ferreira VC, et al. (2014) The role of lipotoxicity in smoke cardiomyopathy. PLoS One 9: e113739. doi: 10.1371/journal.pone.0113739
|
| [71] | Zhang Y, Hai J, Cao M, et al. (2013) Silibinin ameliorates steatosis and insulin resistance during non-alcoholic fatty liver disease development partly through targeting IRS-1/PI3K/Akt pathway. Int Immunopharmacol 17: 714-720. |
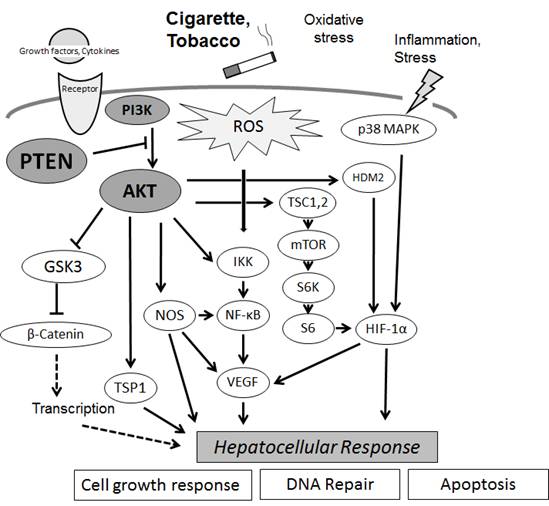
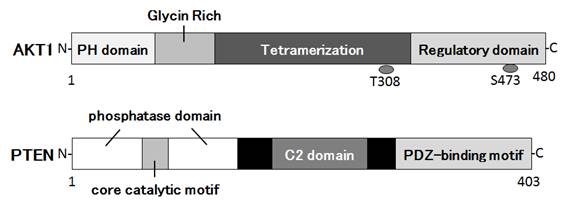
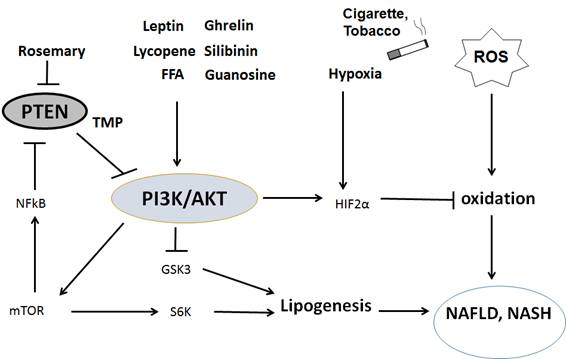
Figures(3)

Mayuko Ichimura, Akari Minami, Noriko Nakano, Yasuko Kitagishi, Toshiyuki Murai, Satoru Matsuda. Cigarette smoke may be an exacerbation factor in nonalcoholic fatty liver disease via modulation of the PI3K/AKT pathway[J]. AIMS Molecular Science, 2015, 2(4): 427-439. doi: 10.3934/molsci.2015.4.427







 DownLoad:
DownLoad: