Citation: Jin Hwan Do. Genome-wide transcriptional comparison of MPP+ treated human neuroblastoma cells with the state space model[J]. AIMS Molecular Science, 2015, 2(4): 440-460. doi: 10.3934/molsci.2015.4.440

| [1] |
Dawson TM, Ko HS, Dawson VL (2010) Genetic animal models of Parkinson's disease. Neuron 66: 646-661. doi: 10.1016/j.neuron.2010.04.034

|
| [2] |
Ribeiro RP, Moreira EL, Santos DB, et al. (2003) Probucol affords neuroprotection in a 6-OHDA mouse model of Parkinson's disease. Neurochem Res 38: 660-668. doi: 10.1007/s11064-012-0965-0
|
| [3] |
Lu C, Zhang J, Shi X, et al. (2014) Neuroprotective effects of tetramethylpyrazine against dopaminergic neuron injury in a rat model of Parkinson's disease induced by MPTP. Int J Biol Sci 10: 350-357. doi: 10.7150/ijbs.8366
|
| [4] |
Langston JW, Ballard P, Tetrud JW, et al. (1983) Parkinsonism in humans due to a product of meperidine- analog synthesis. Science 219: 979-980. doi: 10.1126/science.6823561
|
| [5] |
Dagda RK, Das Banerjee T, Janda E (2013) How Parkinsonian toxins dysregulate the autophagy machinery. Int J Mol Sci 14: 22163-22189. doi: 10.3390/ijms141122163
|
| [6] |
Behl C, Davies JB, Lesley R, et al. (1994) Hydrogen peroxide mediates amyloid protein toxicity. Cell 77: 817-827. doi: 10.1016/0092-8674(94)90131-7
|
| [7] |
Xiang W, Schlachetzki JC, Helling S, et al. (2013) Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci 54: 71-83. doi: 10.1016/j.mcn.2013.01.004
|
| [8] |
Zhou M, Xu S, Mi J, et al. (2013) Nuclear translocation of alpha-synuclein increases susceptibility of MES23.5 cells to oxidative stress. Brain Res 1500: 19-27. doi: 10.1016/j.brainres.2013.01.024
|
| [9] |
Varcin M, Bentea E, Michotte Y, et al. (2012) Oxidative stress in genetic mouse models of Parkinson's disease. Oxid Med Cell Longev 2012: 624925-. doi: 10.1155/2012/624925
|
| [10] |
Perier C, Bové J, Vila M (2012) Mitochondria and programmed cell death in Parkinson's disease: apoptosis and beyond. Antioxid Redox Signal 16: 883-895. doi: 10.1089/ars.2011.4074
|
| [11] |
Chakraborty S, Bornhorst J, Nguyen TT, et al. (2013) Oxidative stress mechanisms underlying Parkinson's disease-associated neurodegeneration in C. elegans. Int J Mol Sci 14: 23103-23128. doi: 10.3390/ijms141123103
|
| [12] |
Kim IS, Choi D.-K., Do J.H. (2013) Genome-wide temporal responses of human neuroblastoma SH-SY5Y cells MPP+ neurotoxicity. BioChip J 7: 247-257. doi: 10.1007/s13206-013-7308-3
|
| [13] |
Do JH (2014) Neurotoxin-induced pathway perturbation in human neuroblastoma SH-EP cells. Mol Cells 37: 672-684. doi: 10.14348/molcells.2014.0173
|
| [14] |
Do JH, Kim IS, Lee JD, et al. (2011) Comparison of genomic profiles in human neuroblastic SH-SY5Y and substrate-adherent SH-EP cells using array comparative genomic hybridization. BioChip J 5: 165-174. doi: 10.1007/s13206-011-5210-4
|
| [15] |
Hirose O, Yoshida R, Imoto S, et al. (2008) Statistical inference of transcriptional module-based gene networks from time course gene expression profiles by using state space models. Bioinformatics 24: 932-942. doi: 10.1093/bioinformatics/btm639
|
| [16] |
Leek JT, Monsen E, Dabney AR, et al. (2006) EDGE: extraction and analysis of differential gene expression. Bioinformatics 22: 507-508. doi: 10.1093/bioinformatics/btk005
|
| [17] |
Eden E, Navon R, Steinfeld I, et al. (2009) Gorilla: a tool for discovery and visualization of enriched GO terms in ranked gene list. BMC Bioinformatics 10: 48. doi: 10.1186/1471-2105-10-48
|
| [18] |
Yamaguchi R, Yoshida R, Imoto S, et al. (2007) Finding module-based gene networks with state-space models. IEEE Signal Proc Mag 24: 37-46. doi: 10.1109/MSP.2007.273053
|
| [19] |
Tamada Y, Yamaguchi R, Imoto S, et al. (2011) SiGN-SSM: open source parallel software for estimating gene networks with state space model. Bioinformatics 27: 1172-1173. doi: 10.1093/bioinformatics/btr078
|
| [20] |
Kovács IA, Palotai R, Szalay MS, et al. (2010) Community landscapes: an integrative approach to determine overlapping network module hierarchy, identify key nodes and predict network dynamics. PLoS One 5: e12528. doi: 10.1371/journal.pone.0012528
|
| [21] |
Wang X, Dalkic E, Wu M, et al. (2008) Gene-module analysis: identification to networks and dynamics. Curr Opin Biotechnol 19: 482-491. doi: 10.1016/j.copbio.2008.07.011
|
| [22] |
Pinho R, Garcia V, Irimia M, et al. (2014) Stability depends on positive autoregulation in Boolean gene regulatory networks. PLoS Comput Biol 10: e1003916. doi: 10.1371/journal.pcbi.1003916
|
| [23] |
Iaccarino I, Marra G, Palombo F, et al (1998) hMSH2 and hMSH6 play distinct roles in mismatch binding and contribute differently to the ATPase activity of hMutSalpha. EMBO J 17: 2677-2686. doi: 10.1093/emboj/17.9.2677
|
| [24] |
Ardley HC, Robinson PA (2005) E3 ubiquitin ligases. Essays Biochem. 41: 15-30. doi: 10.1042/bse0410015
|
| [25] | Wu DC, Jackson-Lewis V, Vila M, et al. (2002) Blockage of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22: 1763-1771. |
| [26] |
Perier C, Tieu K, Guégan C, et al. (2005) Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci USA 102: 19126-19131. doi: 10.1073/pnas.0508215102
|
| [27] |
Hoang T, Choi DK, Nagai M, et al. (2009) Neuronal NOS and cyclooxygenase-2 contribute to DNA damage in a mouse model of Parkinson disease. Free Radic Biol Med 47: 1049-1056. doi: 10.1016/j.freeradbiomed.2009.07.013
|
| [28] |
Shinohara T, Nagashima K, Major EO (1997) Propagation of the human polyomavirus, JCV, in human neuroblastoma cell lines. Virology 228: 269-277. doi: 10.1006/viro.1996.8409
|
| [29] |
Hondele M, Ladurner AG (2011) The chaperone-histone partnership: for the greater good of histone traffic and chromatin plasticity. Curr Opin Struct Biol 21: 698–708. doi: 10.1016/j.sbi.2011.10.003
|
| [30] |
Smith KT, Workman JL (2012) Chromatin proteins: key responders to stress. PLoS Biol 10: e1001371. doi: 10.1371/journal.pbio.1001371
|

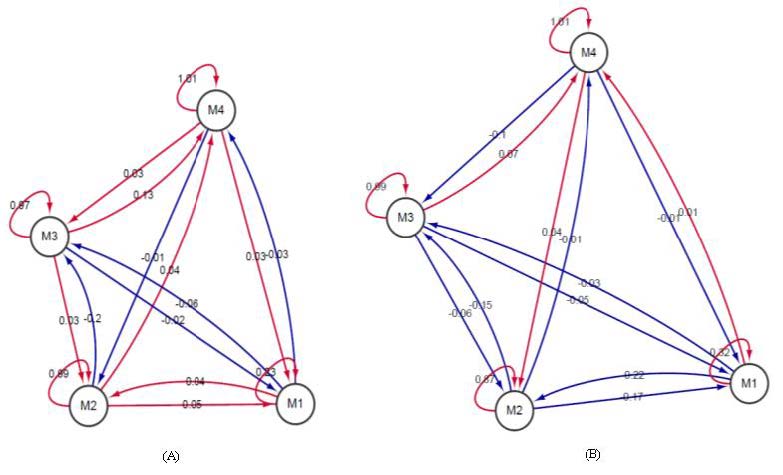
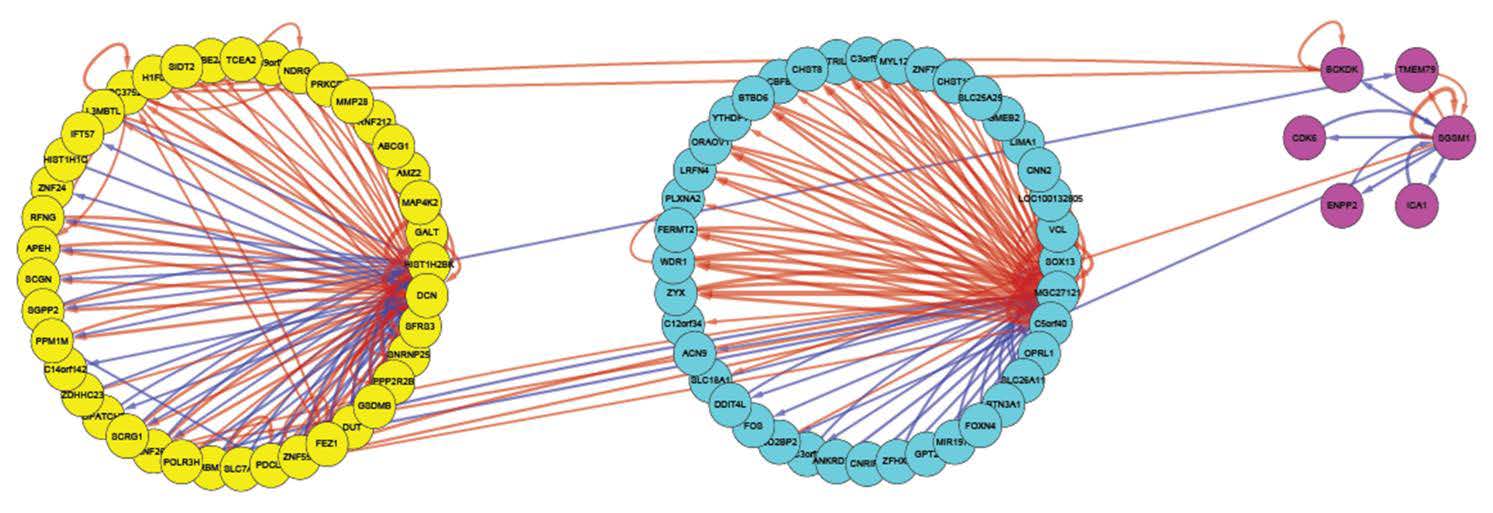
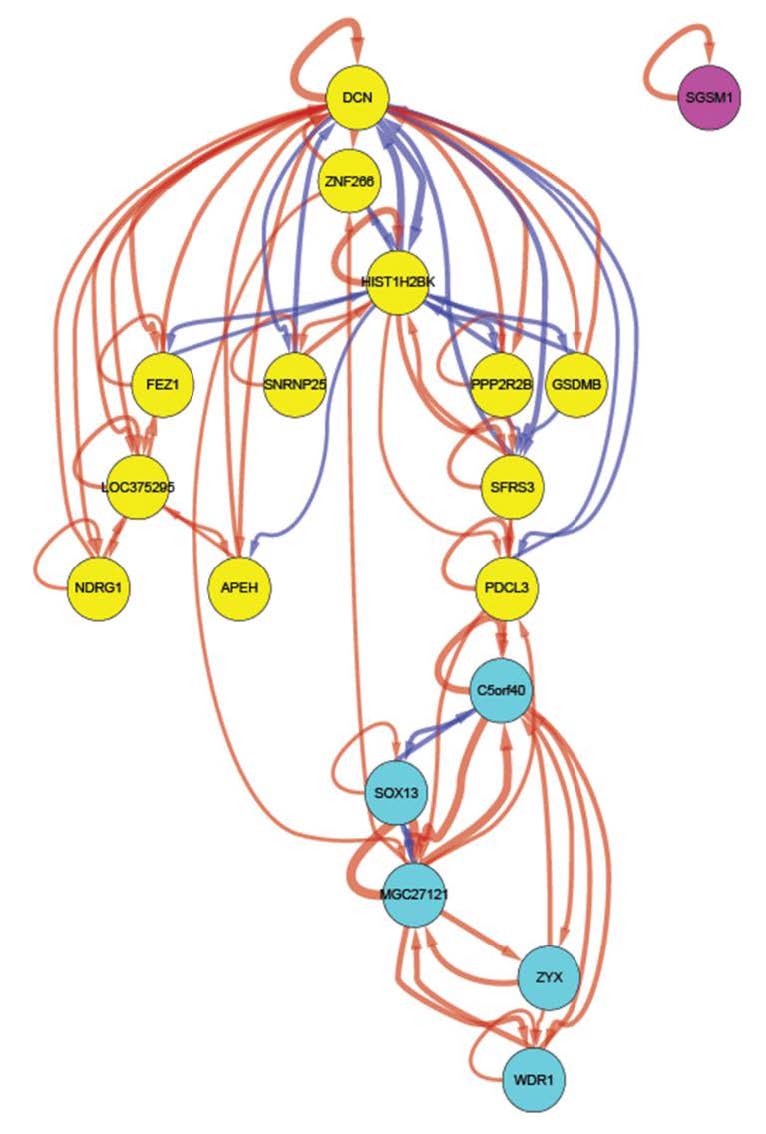
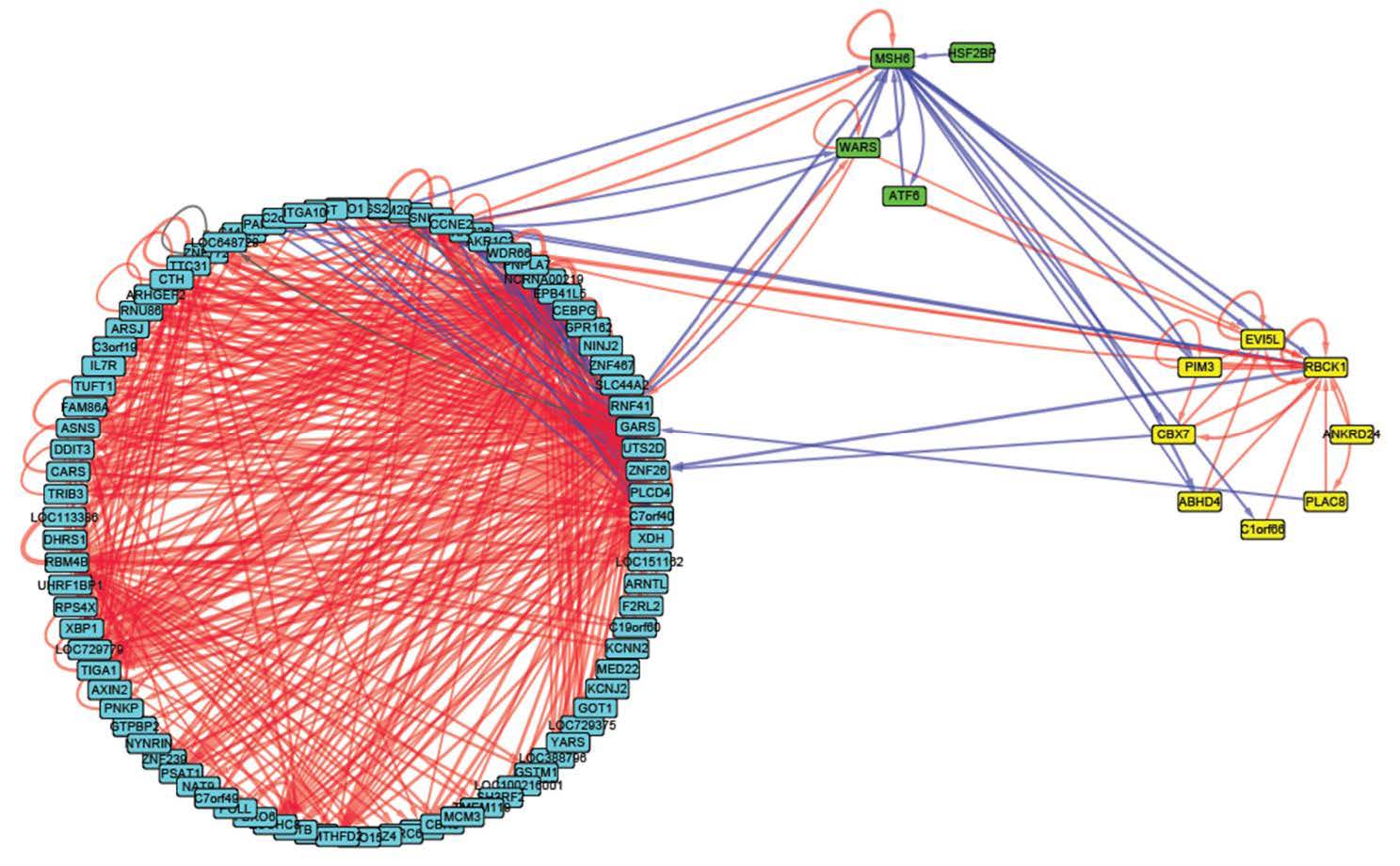
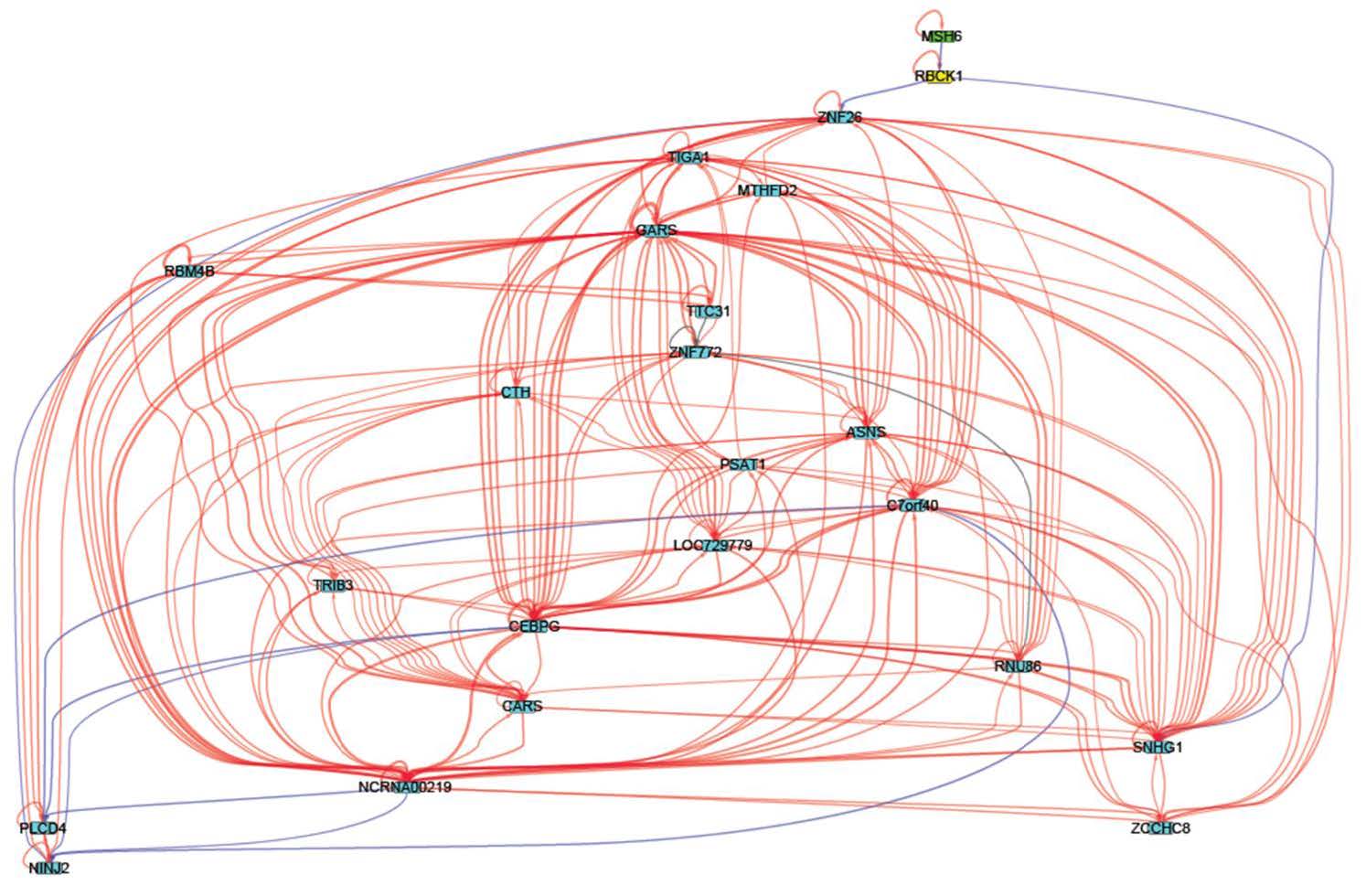
Figures(6) / Tables(3)

Jin Hwan Do. Genome-wide transcriptional comparison of MPP+ treated human neuroblastoma cells with the state space model[J]. AIMS Molecular Science, 2015, 2(4): 440-460. doi: 10.3934/molsci.2015.4.440







 DownLoad:
DownLoad: