Citation: Benoît H. Lessard, Timothy P. Bender. Controlled and selective placement of boron subphthalocyanines on either chain end of polymers synthesized by nitroxide mediated polymerization[J]. AIMS Molecular Science, 2015, 2(4): 411-426. doi: 10.3934/molsci.2015.4.411

| [1] |
Claessens CG, González-Rodríguez D, Rodríguez-Morgade MS, et al. (2014) Subphthalocyanines, Subporphyrazines, and Subporphyrins: Singular Nonplanar Aromatic Systems. Chem Rev 114: 2192-2277 doi: 10.1021/cr400088w

|
| [2] |
Morse GE, Bender TP (2012) Boronsubphthalocyanines as Organic Electronic Materials. ACS Appl Mater Interfaces 4: 5055-5068. doi: 10.1021/am3015197
|
| [3] |
Mauldin CE, Piliego C, Poulsen D, et al. (2010) Axial Thiophene-Boron(subphthalocyanine) Dyads and Their Application in Organic Photovoltaics. ACS Appl Mater Interfaces 2: 2833-2838. doi: 10.1021/am100516a
|
| [4] |
Kulshreshtha C, Kim GW, Lampande R (2013) New interfacial materials for rapid hole-extraction in organic photovoltaic cells. J Mater Chem 1: 4077-4082. doi: 10.1039/c3ta00808h
|
| [5] |
Yasuda T, Tsutsui T (2006) n-Channel Organic Field-Effect Transistors Based on Boron-Subphthalocyanine. Mol Cryst Liq Cryst 462: 3-9. doi: 10.1080/15421400601009278
|
| [6] |
Melville O, Lessard BH, Bender TP (2015) Phthalocyanine Based Organic Thin-Film Transistors: A Review of Recent Advances. ACS Appl Mater Interfaces 7: 13105-13118. doi: 10.1021/acsami.5b01718
|
| [7] |
Morse GE, Castrucci JS, Helander MG, et al. (2011) Phthalimido-boronsubphthalocyanines: New Derivatives of Boronsubphthalocyanine with Bipolar Electrochemistry and Functionality in OLEDs. ACS Appl Mater Interfaces 3: 3538-3544. doi: 10.1021/am200758w
|
| [8] | Dang JD, Virdo JD, Lessard BH, et al. (2012) A Boron Subphthalocyanine Polymer: Poly(4-methylstyrene)- co-poly(phenoxy boron subphthalocyanine). Macromolecules 45: 7791-7798. |
| [9] | Lessard BH, Bender TP (2013) Boron Subphthalocyanine Polymers by Facile Coupling to Poly(acrylic acid-ran-styrene) Copolymers Synthesized by Nitroxide-Mediated Polymerization and the Associated Problems with Autoinitiation. Macromol Rapid Commun 34: 568-573. |
| [10] | Lessard BH, Sampson KL, Plint T, et al. (2015) Boron subphthalocyanine polymers: Avoiding the small molecule side product and exploring their use in organic light-emitting diodes. J Polym Sci Pol Chem 53: 1996-2006. |
| [11] | Kim Y, Cook S, Kirkpatrick J, et al. (2007) Effect of the End Group of Regioregular Poly(3-hexylthiophene) Polymers on the Performance of Polymer/Fullerene Solar Cells. J Phys Chem C 111: 8137-8141. |
| [12] |
Handa NV, Serrano AV, Robb MJ, et al. (2015) Exploring the synthesis and impact of end-functional poly(3-hexylthiophene). J Polym Sci Pol Chem 53: 831-841. doi: 10.1002/pola.27522
|
| [13] |
Wang Q, Zhang B, Liu L, et al. (2012) Effect of End Groups on Optoelectronic Properties of Poly(9,9-dioctylfluorene): A Study with Hexadecylfluorenes as Model Polymers. J Phys Chem C 116: 21727-21733. doi: 10.1021/jp3083369
|
| [14] | Nicolas J, Guillaneuf Y, Lefay C, et al. (2012) Nitroxide-Mediated Polymerization. Prog Polym Sci 38: 63-235. |
| [15] |
Grubbs R (2011) Nitroxide-Mediated Radical Polymerization: Limitations and Versatility. Polym Rev 51: 104-137. doi: 10.1080/15583724.2011.566405
|
| [16] |
Hawker CJ, Bosman A, Harth E (2001) New polymer synthesis by nitroxide mediated living radical polymerizations. Chem Rev 101: 3661-3688. doi: 10.1021/cr990119u
|
| [17] |
Georges M, Veregin R, Kazmaier P (1993) Narrow molecular weight resins by a free-radical polymerization process. Macromolecules 26: 2987-2988. doi: 10.1021/ma00063a054
|
| [18] |
Benoit D, Grimaldi S, Robin S, et al. (2000) Kinetics and mechanism of controlled free-radical polymerization of styrene and n-butyl acrylate in the presence of an acyclic b-phosphonylated nitroxide. J Am Chem Soc 122: 5929-5939. doi: 10.1021/ja991735a
|
| [19] |
Benoit D, Chaplinski V, Braslau R, et al. (1999) Development of a universal alkoxyamine for “living” free radical polymerizations. J Am Chem Soc 121: 3904-3920. doi: 10.1021/ja984013c
|
| [20] |
Eggenhuisen TM, Remzi Becer C, Fijten MWM, et al. (2008) Libraries of statistical hydroxypropyl acrylate containing copolymers with LCST properties prepared by NMP. Macromolecules 41: 5132-5140. doi: 10.1021/ma800469p
|
| [21] | Lessard BH, Tervo C, Maric M (2009) High-Molecular-Weight Poly(tert-butyl acrylate) by Nitroxide-Mediated Polymerization: Effect of Chain Transfer to Solvent. Macromol React Eng 3: 245-256. |
| [22] | Savelyeva X, Lessard B (2012) Amphiphilic Poly (4‐acryloylmorpholine)/Poly [2‐(N‐carbazolyl) ethyl acrylate] Random and Block Copolymers Synthesized by NMP. Macromol React Eng 6: 200-212. |
| [23] |
Nicolas J, Brusseau S, Charleux B (2010) A minimal amount of acrylonitrile turns the nitroxide-mediated polymerization of methyl methacrylate into an almost ideal controlled/living system. J Polym Sci Pol Chem 48: 34-47. doi: 10.1002/pola.23749
|
| [24] |
Lessard BH, Guillaneuf Y, Mathew M, et al. (2013) Understanding the Controlled Polymerization of Methyl Methacrylate with Low Concentrations of 9-(4-Vinylbenzyl)-9H-carbazole Comonomer by Nitroxide-Mediated Polymerization: The Pivotal Role of Reactivity Ratios. Macromolecules 46: 805-813. doi: 10.1021/ma3023525
|
| [25] |
Lessard BH, Ling EJY, Morin MST, et al. (2011) Nitroxide-mediated radical copolymerization of methyl methacrylate controlled with a minimal amount of 9-(4-vinylbenzyl)-9H-carbazole. J Polym Sci Pol Chem 49: 1033-1045. doi: 10.1002/pola.24522
|
| [26] | Frank B, Gast AP, Russell TP, et al. (1996) Polymer Mobility in Thin Films. Macromolecules 29: 6531-6534. |
| [27] | Mather BD, Lizotte JR, Long TE (2004) Synthesis of Chain End Functionalized Multiple Hydrogen Bonded Polystyrenes and Poly(alkyl acrylates) Using Controlled Radical Polymerization. Macromolecules 37: 9331-9337. |
| [28] |
Chauvin F, Dufils P-E, Gigmes D, et al. (2006) Nitroxide-Mediated Polymerization: The Pivotal Role of the kd Value of the Initiating Alkoxyamine and the Importance of the Experimental Conditions. Macromolecules 39: 5238-5250. doi: 10.1021/ma0527193
|
| [29] |
Lessard BH, Maric M (2010) One-Step Poly(styrene-alt-maleic anhydride)-block-poly(styrene) Copolymers with Highly Alternating Styrene/Maleic Anhydride Sequences Are Possible by Nitroxide-Mediated Polymerization. Macromolecules 43: 879-885. doi: 10.1021/ma902234t
|
| [30] |
Lessard BH, Maric M (2008) Effect of an Acid Protecting Group on the “Livingness” of Poly(acrylic acid-ran-styrene) Random Copolymer Macroinitiators for Nitroxide-Mediated Polymerization of Styrene. Macromolecules 41: 7881-7891. doi: 10.1021/ma801255g
|
| [31] | Lessard BH, Aumand-Bourque C, Chaudury R, et al. (2011) Poly(ethylene-co-butylene)-b-(styrene-ran-maleic anhydride)2 Compatibilizers via Nitroxide Mediated Radical Polymerization. Int Polym Process XXVI: 197-204. |
| [32] |
Dufils P-E, Chagneux N, Gigmes D, et al. (2007) Intermolecular radical addition of alkoxyamines onto olefins: An easy access to advanced macromolecular architectures precursors. Polymer 48: 5219-5225. doi: 10.1016/j.polymer.2007.06.050
|
| [33] |
Gigmes D, Dufils P-E, Glé D, et al. (2011) Intermolecular radical 1,2-addition of the BlocBuilder MA alkoxyamine onto activated olefins: a versatile tool for the synthesis of complex macromolecular architecture. Polym Chem 2: 1624-1631. doi: 10.1039/c1py00057h
|
| [34] |
Chenal M, Boursier C, Guillaneuf Y, et al. (2011) First peptide/protein PEGylation with functional polymers designed by nitroxide-mediated polymerization. Polym Chem 2: 1523-1530. doi: 10.1039/c1py00028d
|
| [35] | Parvole J, Ahrens L, Blas H, et al. (2009) Grafting polymer chains bearing an N-succinimidyl activated ester end-group onto primary amine-coated silica particles and application of a simple, one-step approach via nitroxide-mediated controlled/living free-radical polymerization. J Polym Sci Pol Chem 48: 173-185. |
| [36] |
Petit C, Luneau B, Beaudoin E, et al. (2007) Liquid chromatography at the critical conditions in pure eluent: An efficient tool for the characterization of functional polystyrenes. J Chromatography A 1163: 128-137. doi: 10.1016/j.chroma.2007.06.039
|
| [37] |
Harth E, Hawker C, Fan W (2001) Chain end functionalization in nitroxide-mediated “living” free radical polymerizations. Macromolecules 34: 3856-3862. doi: 10.1021/ma0019297
|
| [38] | Turro NJ, Lem G, Zavarine IS (2000) A living free radical exchange reaction for the preparation of photoactive end-labeled monodisperse polymers. Macromolecules 33: 9782-9785. |
| [39] |
Ballesteros OG, Maretti L, Sastre R, et al. (2001) Kinetics of Cap Separation in Nitroxide-Regulated “Living” Free Radical Polymerization: Application of a Novel Methodology Involving a Prefluorescent Nitroxide Switch. Macromolecules 34: 6184-6187. doi: 10.1021/ma0103831
|
| [40] |
Jhaveri SB, Beinhoff M, Hawker CJ, et al. (2008) Chain-End Functionalized Nanopatterned Polymer Brushes Grown viain SituNitroxide Free Radical Exchange. ACS Nano 2: 719-727. doi: 10.1021/nn8000092
|
| [41] |
Guillaneuf Y, Dufils P-E, Autissier L, et al. (2010) Radical Chain End Chemical Transformation of SG1-Based Polystyrenes. Macromolecules 43: 91-100. doi: 10.1021/ma901838m
|
| [42] |
Fulford MV, Jaidka D, Paton AS, et al. (2012) Crystal Structures, Reaction Rates, and Selected Physical Properties of Halo-Boronsubphthalocyanines (Halo = Fluoride, Chloride, and Bromide). J Chem Eng Data 57: 2756-2765. doi: 10.1021/je3005112
|
| [43] | Bruker (2007) APEX2, SAINT & SADABS. Madison, Wisconsin, USA. |
| [44] | Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr A A64: 112-122. |
| [45] | Morse GE, Paton AS, Lough A, et al. (2010) Chloro boron subphthalocyanine and its derivatives: dyes, pigments or somewhere in between? Dalton Transactions 39: 3915-3922. |
| [46] |
Beija M, Charreyre MT, Martinho J (2011) Dye-labelled polymer chains at specific sites: Synthesis by living/controlled polymerization. Prog Polym Sci 36: 568-602. doi: 10.1016/j.progpolymsci.2010.06.004
|
| [47] |
Scott ME, Parent JS, Hennigar SL, et al. (2002) Determination of Alkoxyamine Concentrations in Nitroxyl-Mediated Styrene Polymerization Products. Macromolecules 35: 7628-7633. doi: 10.1021/ma020679m
|
| [48] |
Zetterlund P, Saka Y, McHale R, et al. (2006) Nitroxide-mediated radical polymerization of styrene: Experimental evidence of chain transfer to monomer. Polymer 47: 7900-7908. doi: 10.1016/j.polymer.2006.09.033
|
| [49] |
Asua JM, Beuermann S, Buback M, et al. (2004) Critically Evaluated Rate Coefficients for Free-Radical Polymerization, 5. Macromol Chem Phys 205: 2151-2160. doi: 10.1002/macp.200400355
|
| [50] | Hui AW, Hamielec AE (1978) Thermal polymerization of styrene. J App Polym Sci 22: 1207-1223. |
| [51] |
Hui AW, Hamielec AE (1972) Thermal polymerization of styrene at high conversions and temperatures. An experimental study. J App Polym Sci 16: 749-769. doi: 10.1002/app.1972.070160319
|
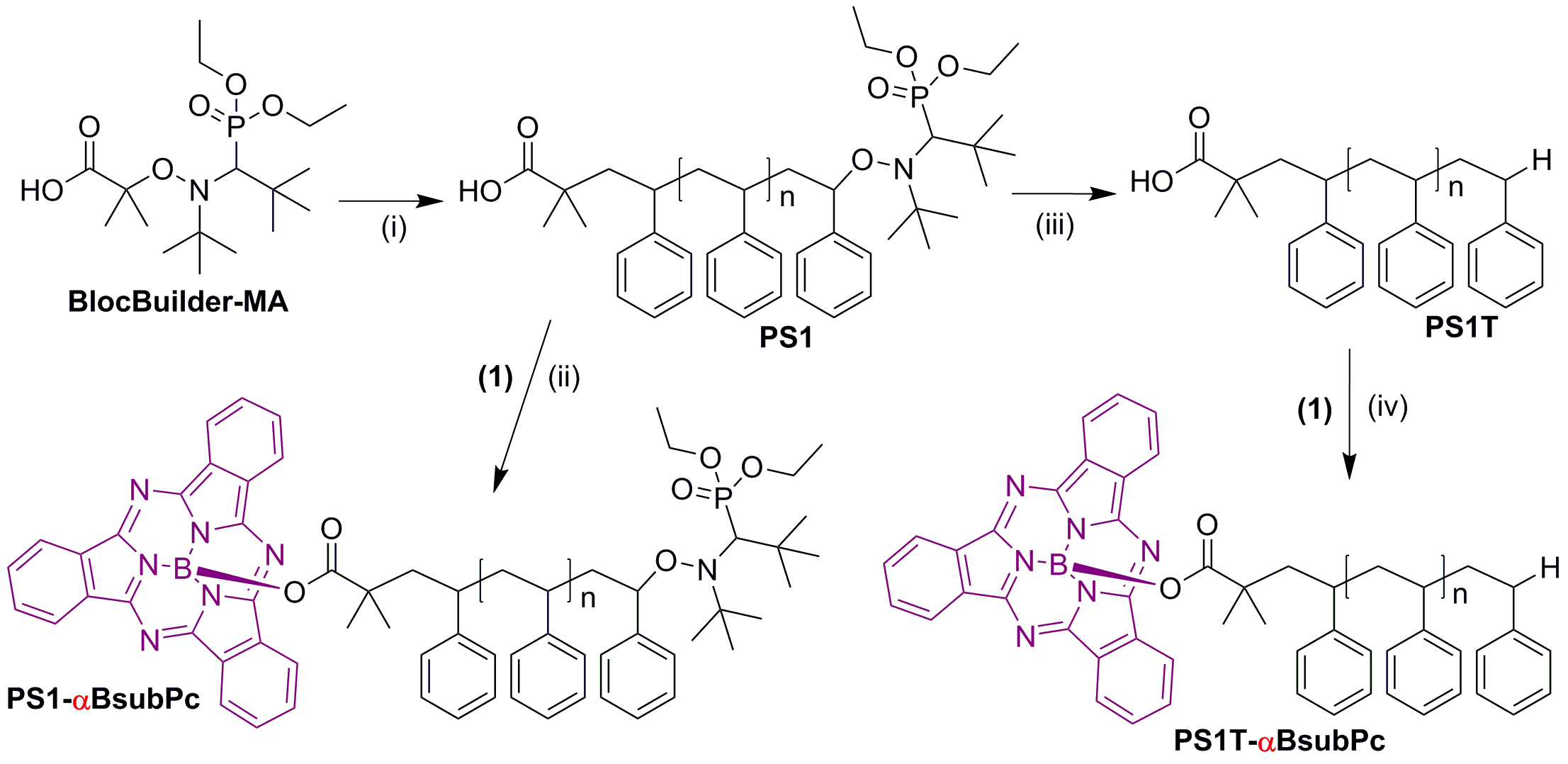
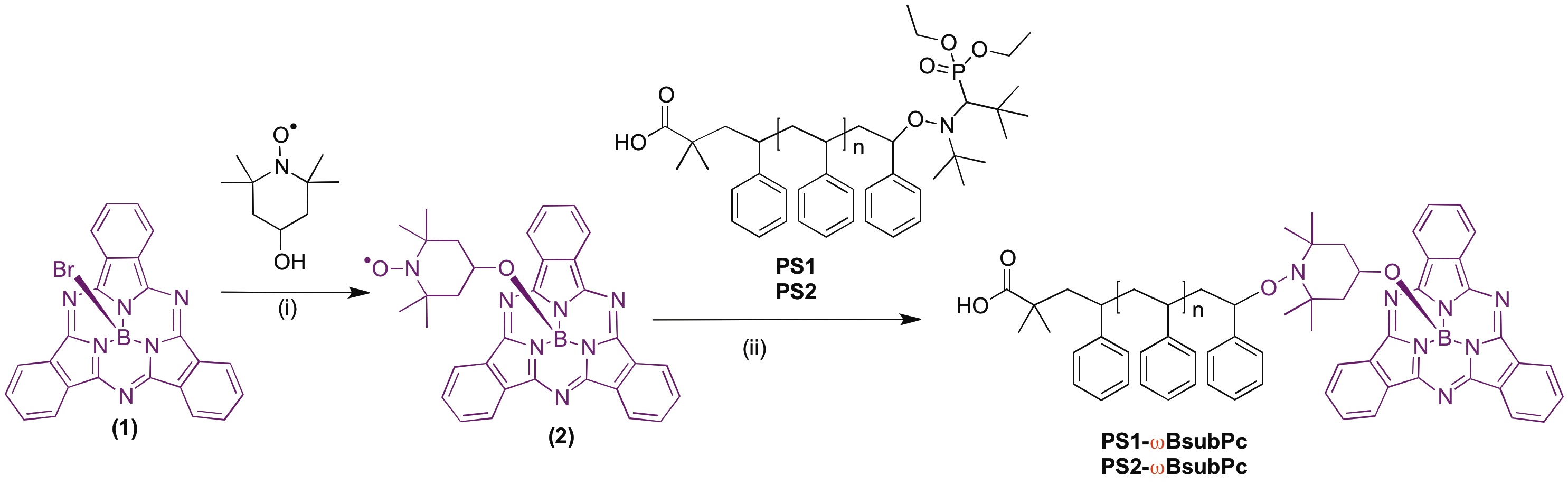
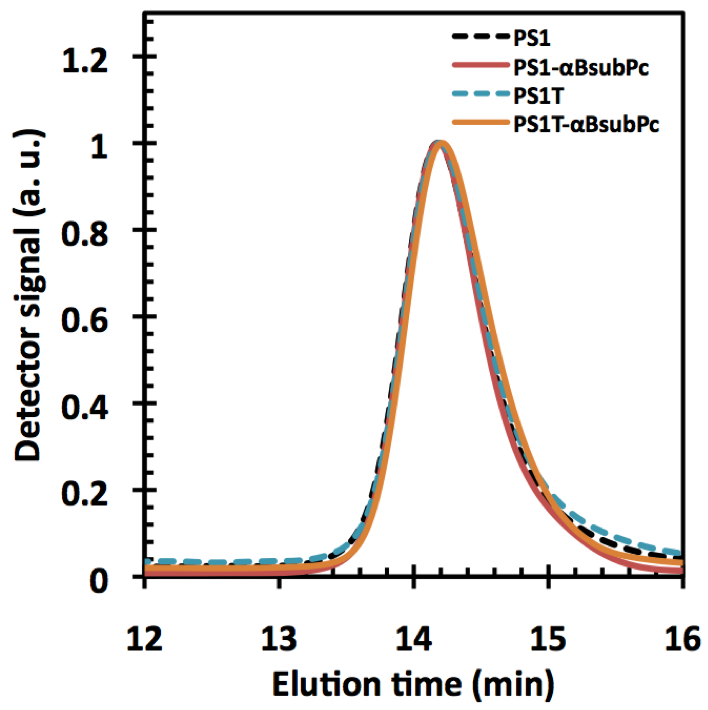
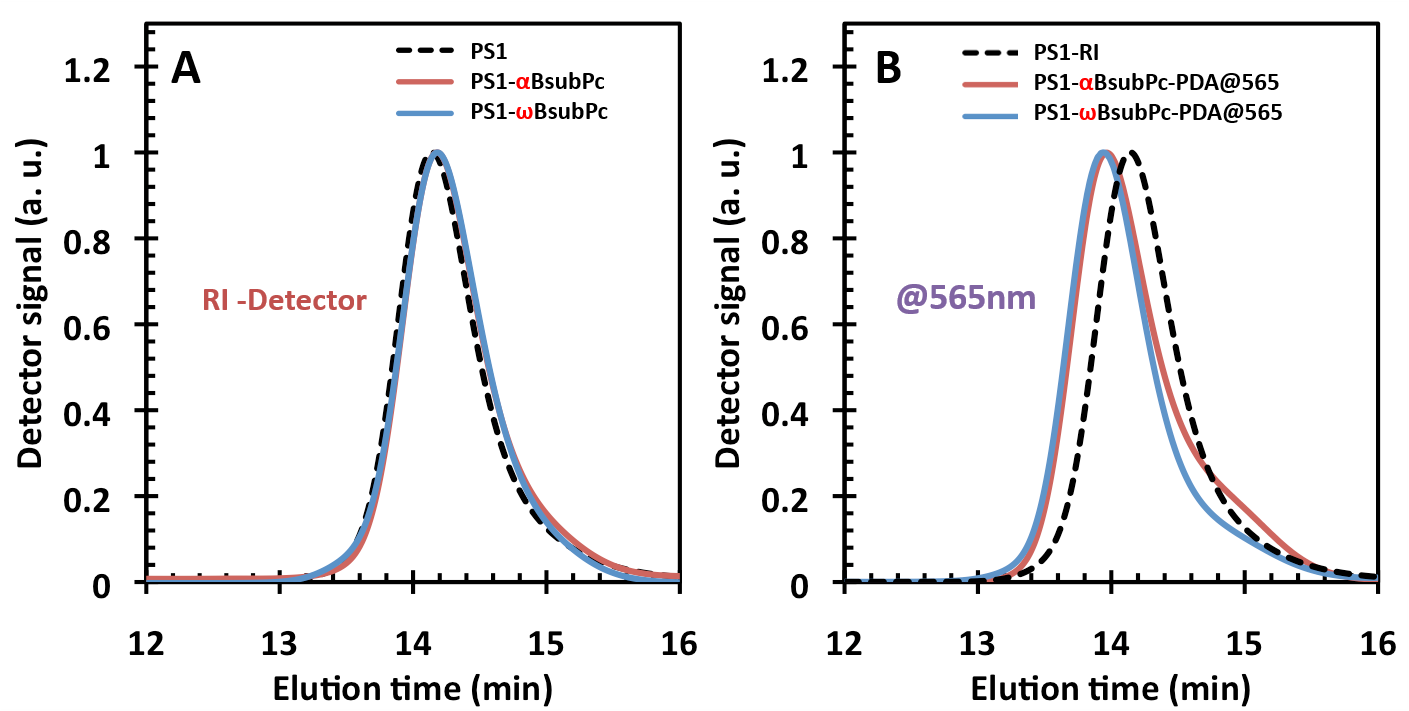
Figures(8) / Tables(1)

Benoît H. Lessard, Timothy P. Bender. Controlled and selective placement of boron subphthalocyanines on either chain end of polymers synthesized by nitroxide mediated polymerization[J]. AIMS Molecular Science, 2015, 2(4): 411-426. doi: 10.3934/molsci.2015.4.411







 DownLoad:
DownLoad: