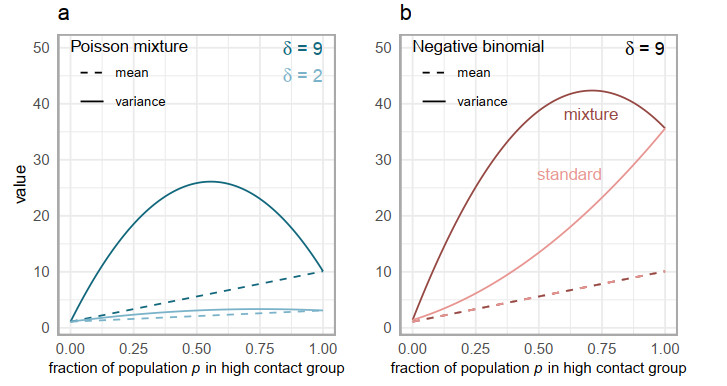
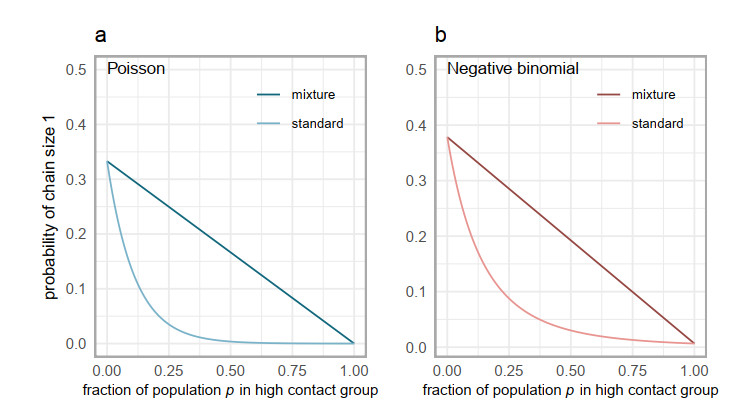
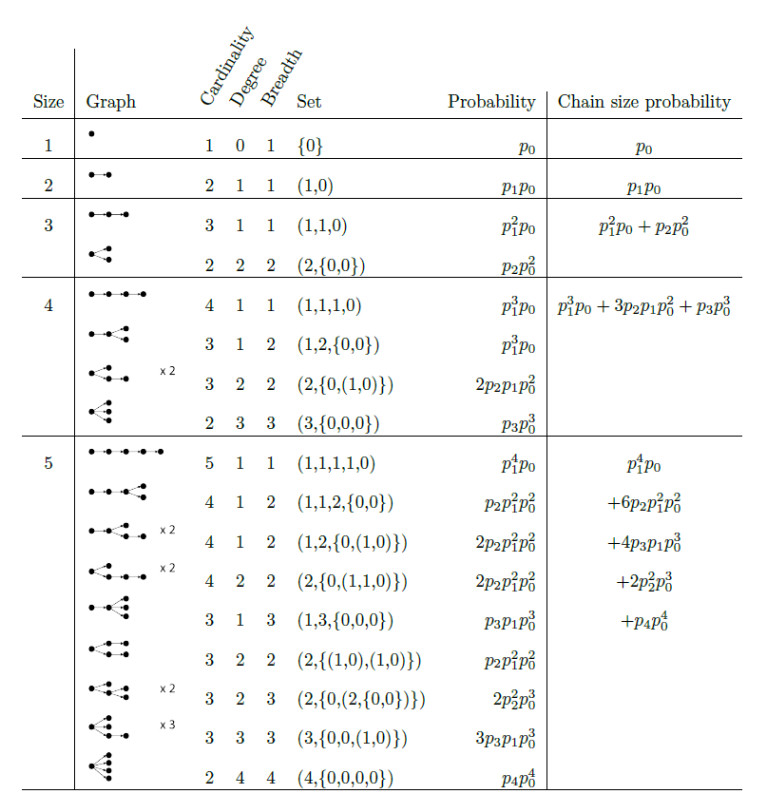
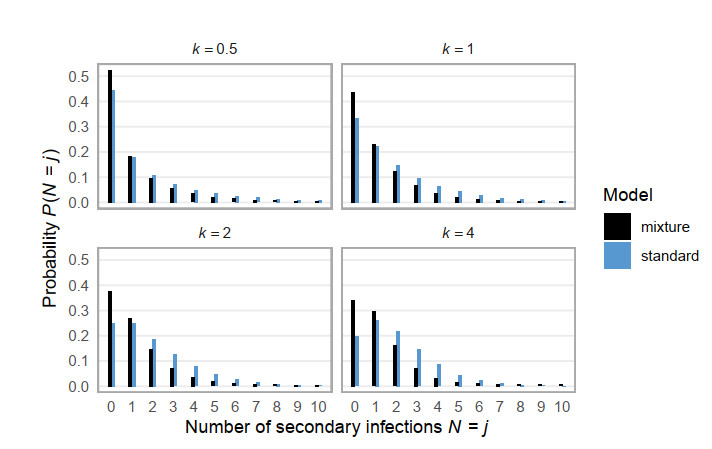
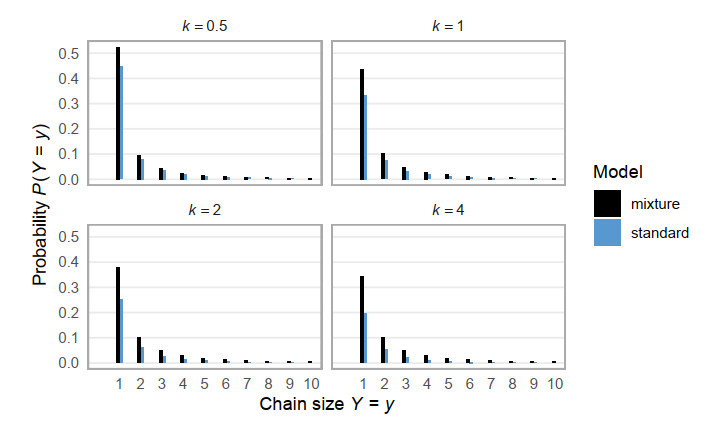
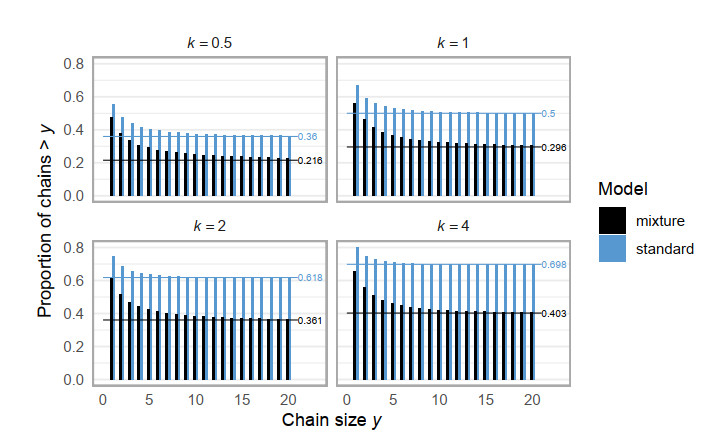
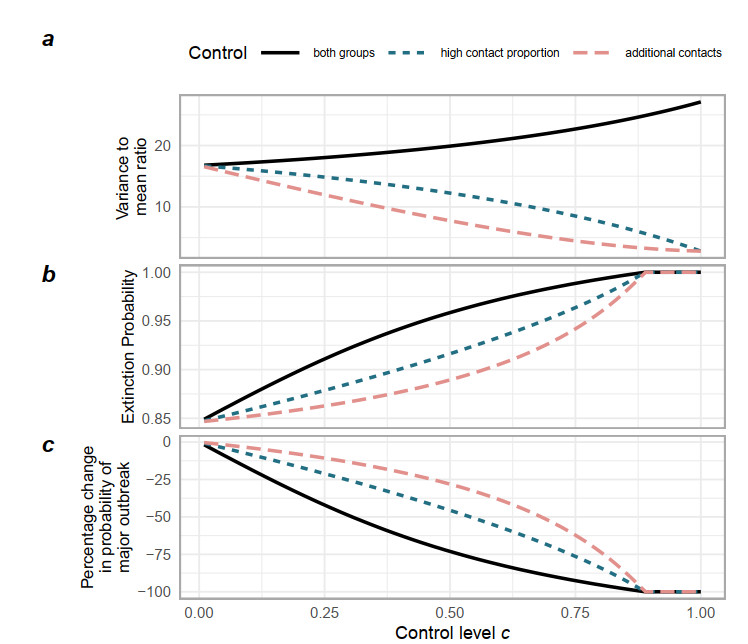
Superspreading transmission is usually modeled using the negative binomial distribution, simply because its variance is larger than the mean and it can be long-tailed. However, populations are often partitioned into groups by social, behavioral, or environmental risk factors, particularly in closed settings such as workplaces or care homes. While heterogeneities in infectious histories and contact structure have been considered separately, models for superspreading events that include the joint effects of social and biological risk factors are lacking. To address this need, we developed a mechanistic finite mixture model for the number of secondary infections that unites population partitioning with individual-level heterogeneity in infectious period duration. We showed that the variance in the number of secondary infections is composed of both sources of heterogeneity: risk group structuring and infectiousness. We used the model to construct the outbreak size distribution and to derive critical thresholds for elimination resulting from control activities that differentially target the high-contact subpopulation vs. the population at large. We compared our model with the standard negative binomial distribution and showed that the tail behavior of the outbreak size distribution under a finite mixture model differs substantially. Our results indicate that even if the infectious period follows a bell-shaped distribution, heterogeneity in outbreak sizes may arise due to the influence of population risk structure.
Citation: Suzanne M. O'Regan, John M. Drake. Finite mixture models of superspreading in epidemics[J]. Mathematical Biosciences and Engineering, 2025, 22(5): 1081-1108. doi: 10.3934/mbe.2025039
Superspreading transmission is usually modeled using the negative binomial distribution, simply because its variance is larger than the mean and it can be long-tailed. However, populations are often partitioned into groups by social, behavioral, or environmental risk factors, particularly in closed settings such as workplaces or care homes. While heterogeneities in infectious histories and contact structure have been considered separately, models for superspreading events that include the joint effects of social and biological risk factors are lacking. To address this need, we developed a mechanistic finite mixture model for the number of secondary infections that unites population partitioning with individual-level heterogeneity in infectious period duration. We showed that the variance in the number of secondary infections is composed of both sources of heterogeneity: risk group structuring and infectiousness. We used the model to construct the outbreak size distribution and to derive critical thresholds for elimination resulting from control activities that differentially target the high-contact subpopulation vs. the population at large. We compared our model with the standard negative binomial distribution and showed that the tail behavior of the outbreak size distribution under a finite mixture model differs substantially. Our results indicate that even if the infectious period follows a bell-shaped distribution, heterogeneity in outbreak sizes may arise due to the influence of population risk structure.

| [1] |
J. O. Lloyd-Smith, S. J. Schreiber, P. E. Kopp, W. M. Getz, Superspreading and the effect of individual variation on disease emergence, Nature, 438 (2005), 355–359. https://doi.org/10.1038/nature04153 doi: 10.1038/nature04153

|
| [2] |
S. Funk, M. Salathé, V. A. A. Jansen, Modelling the influence of human behaviour on the spread of infectious diseases: a review, J. R. Soc. Interface, 7 (2010), 1247–1256. https://doi.org/10.1098/rsif.2010.0142 doi: 10.1098/rsif.2010.0142
|
| [3] |
B. M. Althouse, E. A. Wenger, J. C. Miller, S. V. Scarpino, A. Allard, L. Hébert-Dufresne, et al., Superspreading events in the transmission dynamics of SARS-CoV-2: opportunities for interventions and control, PLoS Biol., 18 (2020), e3000897. https://doi.org/10.1371/journal.pbio.3000897 doi: 10.1371/journal.pbio.3000897
|
| [4] |
A. Endo, H. Murayama, S. Abbott, R. Ratnayake, C. A. B. Pearson, W. J. Edmunds, et al., Heavy-tailed sexual contact networks and monkeypox epidemiology in the global outbreak, Science, 378 (2022), 90–94. https://doi.org/10.1126/science.add4507 doi: 10.1126/science.add4507
|
| [5] |
L. Hamner, P. Dubbel, I. Capron, A. Ross, A. Jordan, J. Lee, et al., High SARS-CoV-2 attack rate following exposure at a choir practice — Skagit County, Washington, March 2020, MMWR Morb. Mortal. Wkly. Rep., 69 (2020), 606–610. https://doi.org/10.15585/mmwr.mm6919e6 doi: 10.15585/mmwr.mm6919e6
|
| [6] |
D. C. Adam, P. Wu, J. Y. Wong, E. H. Y. Lau, T. K. Tsang, S. Cauchemez, et al., Clustering and superspreading potential of SARS-CoV-2 infections in Hong Kong, Nat. Med., 26 (2020), 1714–1719. https://doi.org/10.1038/s41591-020-1092-0 doi: 10.1038/s41591-020-1092-0
|
| [7] | E. Lemieux, K. J. Siddle, B. M. Shaw, C. Loreth, S. F. Schaffner, A. Gladden-Young, et al., Phylogenetic analysis of SARS-CoV-2 in Boston highlights the impact of superspreading events, Science, 371 (2021). https://doi.org/10.1126/science.abe3261 |
| [8] | C. Illingworth, W. L. Hamilton, B. Warne, M. Routledge, A. Popay, C. Jackson, et al., Superspreaders drive the largest outbreaks of hospital onset COVID-19 infections, eLife, 10 (2021). https://doi.org/10.7554/eLife.67308 |
| [9] | C. J. Mode, C. K. Sleeman, Stochastic Processes in Epidemiology: HIV/AIDS, Other Infectious Diseases and Computers, World Scientific Publishing, Singapore, 2000. |
| [10] |
S. Blumberg, J. O. Lloyd-Smith, Inference of R0 and transmission heterogeneity from the size distribution of stuttering chains, PLoS Comput. Biol., 9 (2013), e1002993. https://doi.org/10.1371/journal.pcbi.1002993 doi: 10.1371/journal.pcbi.1002993
|
| [11] | M. J. Keeling, P. Rohani, Modeling Infectious Diseases in Humans and Animals, Princeton University Press, Princeton, 2008. |
| [12] |
K. Rock, S. Brand, J. Moir, M. J. Keeling, Dynamics of infectious diseases, Rep. Prog. Phys., 77 (2014), 026602. https://doi.org/10.1088/0034-4885/77/2/026602 doi: 10.1088/0034-4885/77/2/026602
|
| [13] | K. Sneppen, B. F. Nielsen, R. J. Taylor, L. Simonsen, Overdispersion in COVID-19 increases the effectiveness of limiting nonrepetitive contacts for transmission control, Proc. Natl. Acad. Sci. U.S.A., 118 (2021). https://doi.org/10.1073/pnas.2016623118 |
| [14] |
T. Garske, C. J. Rhodes, The effect of superspreading on epidemic outbreak size distributions, J. Theor. Biol., 253 (2008), 228–237. https://doi.org/10.1016/j.jtbi.2008.02.038 doi: 10.1016/j.jtbi.2008.02.038
|
| [15] | P. Yan, Distribution theory, stochastic processes and infectious disease modelling, in Mathematical Epidemiology (eds. F. Brauer, P. van den Driessche, J. Wu), Springer, Berlin, Heidelberg, (2008), 229–293. |
| [16] | O. Diekmann, H. Heesterbeek, T. Britton, Mathematical Tools for Understanding Infectious Disease Dynamics, Princeton University Press, Princeton, NJ, 2013. |
| [17] |
D. Karlis, E. Xekalaki, Mixed Poisson distributions, Int. Stat. Rev., 73 (2005), 35–58. https://doi.org/10.1111/j.1751-5823.2005.tb00250.x doi: 10.1111/j.1751-5823.2005.tb00250.x
|
| [18] |
D. Anderson, R. Watson, On the spread of a disease with gamma distributed latent and infectious periods, Biometrika, 67 (1980), 191–198. https://doi.org/10.2307/2335333 doi: 10.2307/2335333
|
| [19] |
T. Britton, D. Lindenstrand, Epidemic modelling: aspects where stochasticity matters, Math. Biosci., 222 (2009), 109–116. https://doi.org/10.1016/j.mbs.2009.10.001 doi: 10.1016/j.mbs.2009.10.001
|
| [20] |
A. L. Lloyd, Realistic distributions of infectious periods in epidemic models: changing patterns of persistence and dynamics, Theor. Popul. Biol., 60 (2001), 59–71. https://doi.org/10.1006/tpbi.2001.1525 doi: 10.1006/tpbi.2001.1525
|
| [21] |
H. J. Wearing, P. Rohani, M. J. Keeling, Appropriate models for the management of infectious diseases, PLoS Med., 2 (2005), e174. https://doi.org/10.1371/journal.pmed.0020174 doi: 10.1371/journal.pmed.0020174
|
| [22] |
W. P. Johnson, The curious history of Faà di Bruno's formula, Am. Math. Mon., 109 (2002), 217–234. https://doi.org/10.1080/00029890.2002.11919857 doi: 10.1080/00029890.2002.11919857
|
| [23] |
D. Cvijović, New identities for the partial Bell polynomials, Appl. Math. Lett., 24 (2011), 1544–1547. https://doi.org/10.1016/j.aml.2011.03.043 doi: 10.1016/j.aml.2011.03.043
|
| [24] | T. Rajala, antiphon/BellB: evaluation of the Bell polynomial, 2019, accessed: 2022-3-22. |
| [25] |
M. Dwass, The total progeny in a branching process and a related random walk, J. Appl. Probab., 6 (1969), 682–686. https://doi.org/10.2307/3212112 doi: 10.2307/3212112
|
| [26] |
J. M. Drake, K. Dahlin, P. Rohani, A. Handel, Five approaches to the suppression of SARS-CoV-2 without intensive social distancing, Proc. Biol. Sci., 288 (2021), 20203074. https://doi.org/10.1098/rspb.2020.3074 doi: 10.1098/rspb.2020.3074
|
| [27] |
C. Kremer, A. Torneri, S. Boesmans, H. Meuwissen, S. Verdonschot, K. V. Driessche, et al., Quantifying superspreading for COVID-19 using Poisson mixture distributions, Sci. Rep., 11 (2021), 14107. https://doi.org/10.1038/s41598-021-93578-x doi: 10.1038/s41598-021-93578-x
|
| [28] |
A. Allard, B. M. Althouse, S. V. Scarpino, L. Hébert-Dufresne, Asymmetric percolation drives a double transition in sexual contact networks, Proc. Natl. Acad. Sci. U.S.A., 114 (2017), 8969–8973. https://doi.org/10.1073/pnas.1703073114 doi: 10.1073/pnas.1703073114
|
| [29] |
A. Allard, C. Moore, S. V. Scarpino, B. M. Althouse, L. Hébert-Dufresne, The role of directionality, heterogeneity, and correlations in epidemic risk and spread, SIAM Rev. Soc. Ind. Appl. Math., 65 (2023), 471–492. https://doi.org/10.1137/20m1383811 doi: 10.1137/20m1383811
|
| [30] | A. Manna, L. Dall'Amico, M. Tizzoni, M. Karsai, N. Perra, Generalized contact matrices allow integrating socioeconomic variables into epidemic models, Sci. Adv., 10 (2024), eadk4606. https://www.science.org/doi/10.1126/sciadv.adk4606 |
| [31] | A. Goyal, D. B. Reeves, E. F. Cardozo-Ojeda, J. T. Schiffer, B. T. Mayer, Viral load and contact heterogeneity predict SARS-CoV-2 transmission and super-spreading events, eLife, 10 (2021). https://doi.org/10.7554/eLife.63537 |
| [32] | P. Z. Chen, N. Bobrovitz, Z. Premji, M. Koopmans, D. N. Fisman, F. X. Gu, Heterogeneity in transmissibility and shedding SARS-CoV-2 via droplets and aerosols, eLife, 10 (2021). https://doi.org/10.7554/eLife.65774 |
| [33] |
C. C. Wang, K. A. Prather, J. Sznitman, J. L. Jimenez, S. S. Lakdawala, Z. Tufekci, et al., Airborne transmission of respiratory viruses, Science, 373 (2021), eabd9149. https://doi.org/10.1126/science.abd9149 doi: 10.1126/science.abd9149
|
Figures(7) / Tables(2)

Suzanne M. O'Regan, John M. Drake. Finite mixture models of superspreading in epidemics[J]. Mathematical Biosciences and Engineering, 2025, 22(5): 1081-1108. doi: 10.3934/mbe.2025039







 DownLoad:
DownLoad: