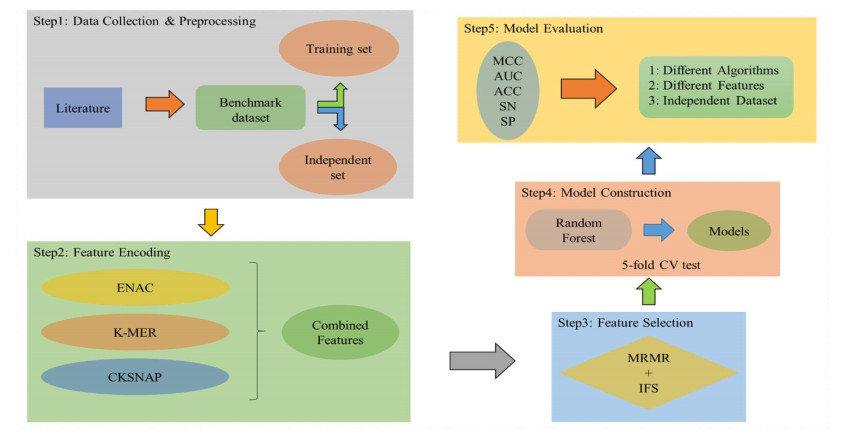
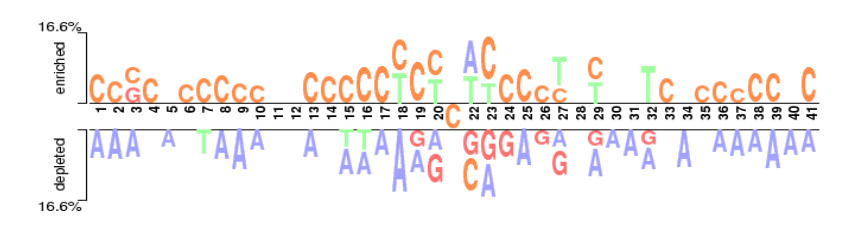
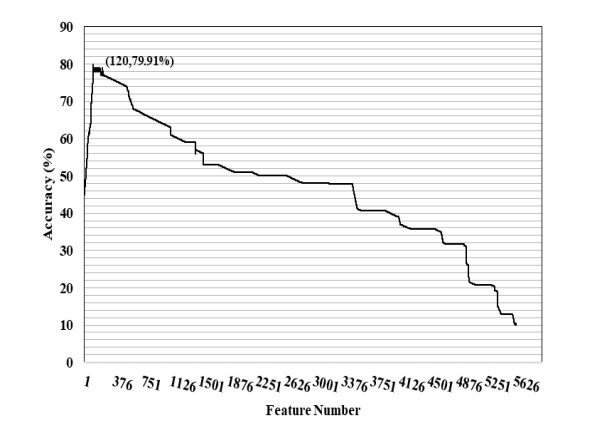
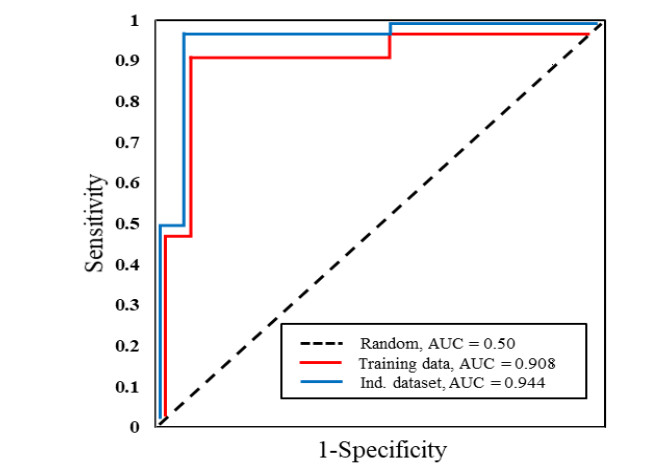
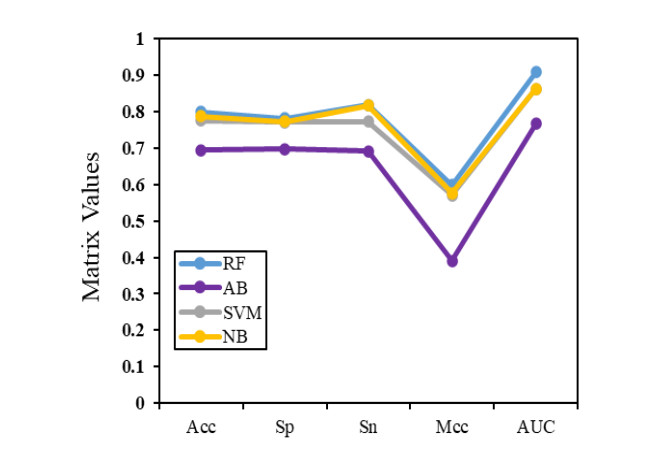
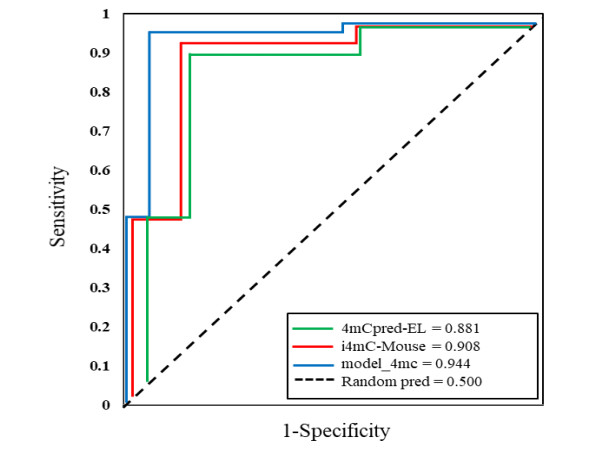
N4-methylcytosine (4mC) is a kind of DNA modification which could regulate multiple biological processes. Correctly identifying 4mC sites in genomic sequences can provide precise knowledge about their genetic roles. This study aimed to develop an ensemble model to predict 4mC sites in the mouse genome. In the proposed model, DNA sequences were encoded by k-mer, enhanced nucleic acid composition and composition of k-spaced nucleic acid pairs. Subsequently, these features were optimized by using minimum redundancy maximum relevance (mRMR) with incremental feature selection (IFS) and five-fold cross-validation. The obtained optimal features were inputted into random forest classifier for discriminating 4mC from non-4mC sites in mouse. On the independent dataset, our model could yield the overall accuracy of 85.41%, which was approximately 3.8% -6.3% higher than the two existing models, i4mC-Mouse and 4mCpred-EL respectively. The data and source code of the model can be freely download from https://github.com/linDing-groups/model_4mc.
Citation: Hasan Zulfiqar, Rida Sarwar Khan, Farwa Hassan, Kyle Hippe, Cassandra Hunt, Hui Ding, Xiao-Ming Song, Renzhi Cao. Computational identification of N4-methylcytosine sites in the mouse genome with machine-learning method[J]. Mathematical Biosciences and Engineering, 2021, 18(4): 3348-3363. doi: 10.3934/mbe.2021167
N4-methylcytosine (4mC) is a kind of DNA modification which could regulate multiple biological processes. Correctly identifying 4mC sites in genomic sequences can provide precise knowledge about their genetic roles. This study aimed to develop an ensemble model to predict 4mC sites in the mouse genome. In the proposed model, DNA sequences were encoded by k-mer, enhanced nucleic acid composition and composition of k-spaced nucleic acid pairs. Subsequently, these features were optimized by using minimum redundancy maximum relevance (mRMR) with incremental feature selection (IFS) and five-fold cross-validation. The obtained optimal features were inputted into random forest classifier for discriminating 4mC from non-4mC sites in mouse. On the independent dataset, our model could yield the overall accuracy of 85.41%, which was approximately 3.8% -6.3% higher than the two existing models, i4mC-Mouse and 4mCpred-EL respectively. The data and source code of the model can be freely download from https://github.com/linDing-groups/model_4mc.

| [1] |
D. Liu, G. Li, Y. Zuo, Function determinants of TET proteins: The arrangements of sequence motifs with specific codes, Brief Bioinform., 20 (2019), 1826-1835. doi: 10.1093/bib/bby053

|
| [2] |
A. Jeltsch, R. Z. Jurkowska, New concepts in DNA methylation, Trends Biochem. Sci., 39 (2014), 310-318. doi: 10.1016/j.tibs.2014.05.002
|
| [3] | D. Schübeler, Function and information content of DNA methylation, Nature, 517 (2015), 321-326. |
| [4] |
B. M. Davis, M. C. Chao, M. K. Waldor, Entering the era of bacterial epigenomics with single molecule real time DNA sequencing, Curr. Opin. Microbiol., 16 (2013), 192-198. doi: 10.1016/j.mib.2013.01.011
|
| [5] |
T. P. Meakin, N. Pillay, S. Beck, 3-methylcytosine in cancer: an underappreciated methyl lesion? Epigenomics, 8 (2016), 451-454. doi: 10.2217/epi.15.121
|
| [6] | K. D. Robertson, DNA methylation and human disease, Nat. Rev. Genet., 6 (2005), 597-610. |
| [7] | M. M. Suzuki, A. Bird, DNA methylation landscapes: provocative insights from epigenomics, Nat. Rev. Genet., 9 (2008), 465-476. |
| [8] |
H. P. Schweizer, Bacterial genetics: Past achievements, present state of the field, and future challenges, Biotechniques, 44 (2008), 633-641. doi: 10.2144/000112807
|
| [9] |
L. M. Iyer, S. Abhiman, L. Aravind, Natural history of eukaryotic DNA methylation systems, Prog. Mol. Biol. Transl. Sci., 101 (2011), 25-104. doi: 10.1016/B978-0-12-387685-0.00002-0
|
| [10] |
W. He, C. Jia, Q. Zou, 4mCPred: Machine learning methods for DNA N4-methylcytosine sites prediction, Bioinformatics, 35 (2019), 593-601. doi: 10.1093/bioinformatics/bty668
|
| [11] |
B. A. Flusberg, D. R. Webster, J. H. Lee, K. J. Travers, E. C. Olivares, T. A. Clark, et al., Direct detection of DNA methylation during single-molecule, real-time sequencing, Nat. Methods, 7 (2010), 461-465. doi: 10.1038/nmeth.1459
|
| [12] | R. Doherty, C. Couldrey, Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: a technical assessment, Front. Genet., 5 (2014), 126. |
| [13] |
J. Boch, U. Bonas, Xanthomonas AvrBs3 family-type III effectors: discovery and function, Annu. Rev. Phytopathol., 48 (2010), 419-436. doi: 10.1146/annurev-phyto-080508-081936
|
| [14] |
W. Chen, H. Yang, P. Feng, H. Ding, H. Lin, iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties, Bioinformatics, 33 (2017), 3518-3523. doi: 10.1093/bioinformatics/btx479
|
| [15] |
L. Wei, R. Su, S. Luan, Z. Liao, B. Manavalan, Q. Zou, et al., Iterative feature representations improve N4-methylcytosine site prediction, Bioinformatics, 35 (2019), 4930-4937. doi: 10.1093/bioinformatics/btz408
|
| [16] |
Z. Lv, D. Wang, H. Ding, B. Zhong, L. Xu, Escherichia coli DNA N-4-methycytosine site prediction accuracy improved by light gradient boosting machine feature selection technology, IEEE Access, 8 (2020), 14851-14859. doi: 10.1109/ACCESS.2020.2966576
|
| [17] |
Q. Tang, J. Kang, J. Yuan, H. Tang, X. Li, H. Lin, et al., DNA4mC-LIP: A linear integration method to identify N4-methylcytosine site in multiple species, Bioinformatics, 36 (2020), 3327-3335. doi: 10.1093/bioinformatics/btaa143
|
| [18] |
B. Manavalan, S. Basith, T. H. Shin, L. Wei, G. Lee, Meta-4mCpred: A sequence-based meta-predictor for accurate DNA 4mC site prediction using effective feature representation, Mol. Ther. Nucleic Acids, 16 (2019), 733-744. doi: 10.1016/j.omtn.2019.04.019
|
| [19] |
B. Manavalan, S. Basith, T. H. Shin, D. Y. Lee, L. Wei, G. Lee, 4mCpred-EL: An ensemble learning framework for identification of DNA N4-methylcytosine sites in the mouse genome, Cells, 8 (2019), 1332. doi: 10.3390/cells8111332
|
| [20] |
M. M. Hasan, B. Manavalan, W. Shoombuatong, M. S. Khatun, H. Kurata, i4mC-Mouse: Improved identification of DNA N4-methylcytosine sites in the mouse genome using multiple encoding schemes, Comput. Struct. Biotechnol. J., 18 (2020), 906-912. doi: 10.1016/j.csbj.2020.04.001
|
| [21] | P. Ye, Y. Luan, K. Chen, Y. Liu, C. Xiao, Z. Xie, MethSMRT: An integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing, Nucleic Acids Res., (2016), DOI: 10.1093/nar/gkw950. |
| [22] | A. Liaw, M. Wiener, Classification and regression by random forest, R. News, 2 (2002), 18-22. |
| [23] |
N. D. Jay, S. P. Cavanagh, C. Olsen, N. E. Hachem, G. Bontempi, B. H. Kains, mRMRe: An R package for parallelized mRMR ensemble feature selection, Bioinformatics, 29 (2013), 2365-2368. doi: 10.1093/bioinformatics/btt383
|
| [24] |
W. Yang, X. J. Zhu, J. Huang, H. Ding, H. Lin, A brief survey of machine learning methods in protein sub-golgi localization, Curr. Bioinform., 14 (2019), 234-240. doi: 10.2174/1574893613666181113131415
|
| [25] |
K. Liu, W. Chen, iMRM: A platform for simultaneously identifying multiple kinds of RNA modifications, Bioinformatics, 36 (2020), 3336-3342. doi: 10.1093/bioinformatics/btaa155
|
| [26] |
L. Fu, B. Niu, Z. Zhu, S. Wu, W. Li, CD-HIT: Accelerated for clustering the next-generation sequencing data, Bioinformatics, 28 (2012), 3150. doi: 10.1093/bioinformatics/bts565
|
| [27] | B. Liu, X. Gao, H. Zhang, BioSeq-Analysis2.0: An updated platform for analyzing DNA, RNA, and protein sequences at sequence level and residue level based on machine learning approaches, Nucleic Acids Res., 47 (2019), e127. |
| [28] | Y. J. Tang, Y. H. Pang, B. Liu, IDP-Seq2Seq: Identification of intrinsically disordered Regions based on sequence to sequence learning, Bioinformaitcs, (2020), DOI: 10.1093/bioinformatics/btaa667. |
| [29] |
N. Schaduangrat, C. Nantasenamat, V. Prachayasittikul, W. Shoombuatong, ACPred: A computational tool for the prediction and analysis of anticancer peptides, Molecules, 24 (2019), 1973. doi: 10.3390/molecules24101973
|
| [30] |
P. Charoenkwan, J. Yana, N. Schaduangrat, C. Nantasenamat, M. M. Hasan, W. Shoombuatong, iBitter-SCM: Identification and characterization of bitter peptides using a scoring card method with propensity scores of dipeptides, Genomics, 112 (2020), 2813-2822. doi: 10.1016/j.ygeno.2020.03.019
|
| [31] |
P. Charoenkwan, C. Nantasenamat, M. M. Hasan, W. Shoombuatong, iTTCA-Hybrid: Improved and robust identification of tumor T cell antigens by utilizing hybrid feature representation, Anal. Biochem., 599 (2020), 113747. doi: 10.1016/j.ab.2020.113747
|
| [32] |
N. Schaduangrat, C. Nantasenamat, V. Prachayasittikul, W. Shoombuatong, Meta-iAVP: A sequence-based meta-predictor for improving the prediction of antiviral peptides using effective feature representation, Int. J. Mol. Sci., 20 (2019), 5743. doi: 10.3390/ijms20225743
|
| [33] |
P. Charoenkwan, C. Nantasenamat, M. M. Hasan, W. Shoombuatong, Meta-iPVP: A sequence-based meta-predictor for improving the prediction of phage virion proteins using effective feature representation, J. Comput. Aided Mol. Des., 34 (2020), 1105-1116. doi: 10.1007/s10822-020-00323-z
|
| [34] |
V. Laengsri, C. Nantasenamat, N. Schaduangrat, P. Nuchnoi, V. Prachayasittikul, W. Shoombuatong, TargetAntiAngio: A sequence-based tool for the prediction and analysis of anti-angiogenic peptides, Int. J. Mol. Sci., 20 (2019), 2950. doi: 10.3390/ijms20122950
|
| [35] |
Y. Zuo, Y. Li, Y. Chen, G. Li, Z. Yan, L. Yang, PseKRAAC: A flexible web server for generating pseudo k-tuple reduced amino acids composition, Bioinformatics., 33 (2017), 122-124. doi: 10.1093/bioinformatics/btw564
|
| [36] | D. Zhang, H. D. Chen, H. Zulfiqar, S. S. Yuan, Q. L. Huang, Z. Y. Zhang, et al., iBLP: An xgboost-based predictor for identifying bioluminescent proteins, Comput. Math. Methods Med., 2021 (2021), 15. |
| [37] | Z. Y. Zhang, Y. H. Yang, H. Ding, D. Wang, W. Chen, H. Lin, Design powerful predictor for mRNA subcellular location prediction in homo sapiens, Brief Bioinform., 22 (2020), 526-535. |
| [38] |
F. Y. Dao, H. Lv, Y. H. Yang, H. Zulfiqar, H. Gao, H. Lin, Computational identification of N6-methyladenosine sites in multiple tissues of mammals, Comput. Struct. Biotechnol. J., 18 (2020), 1084-1091. doi: 10.1016/j.csbj.2020.04.015
|
| [39] |
H. Yang, W. Yang, F. Y. Dao, H. Lv, H. Ding, W. Chen, et al., A comparison and assessment of computational method for identifying recombination hotspots in Saccharomyces cerevisiae, Brief Bioinform., 21 (2020), 1568-1580. doi: 10.1093/bib/bbz123
|
| [40] | L. J. Dou, X. Li, H. Ding, L. Xu, H. Xiang, Is there any sequence feature in the RNA pseudouridine modification prediction problem? Mol. Ther. Nucleic Acids., 19 (2020), 293-303. |
| [41] |
H. Wei, B. Liu, iCircDA-MF: Identification of circRNA-disease associations based on matrix factorization, Brief Bioinform., 21 (2020), 1356-1367. doi: 10.1093/bib/bbz057
|
| [42] | L. Zheng, D. Liu, W. Yang, L. Yang, Y. Zuo, RaacLogo: a new sequence logo generator by using reduced amino acid clusters, Brief Bioinform., (2020), DOI: 10.1093/bib/bbaa096. |
| [43] | H. Lv, F. Y. Dao, H. Zulfiqar, W. Su, H. Ding, L. Liu, et al., A sequence-based deep learning approach to predict CTCF-mediated chromatin loop, Brief Bioinform., (2021), DOI: 10.1093/bib/bbab031. |
| [44] | F. Y. Dao, H. Lv, H. Zulfiqar, H. Yang, W. Su, H. Gao, et al., A computational platform to identify origins of replication sites in eukaryotes, Brief Bioinform., 22 (2020), 1940-1950. |
| [45] |
B. Liu, BioSeq-Analysis: A platform for DNA, RNA, and protein sequence analysis based on machine learning approaches, Brief Bioinform., 20 (2019), 1280-1294. doi: 10.1093/bib/bbx165
|
| [46] | L. Zheng, S. Huang, N. Mu, H. Zhang, J. Zhang, Y. Chang, et al., RAACBook: A web server of reduced amino acid alphabet for sequence-dependent inference by using Chou's five-step rule, Database-Oxford., 2019 (2019), baz131. |
| [47] |
F. Y. Dao, H. Lv, F. Wang, C. Q. Feng, H. Ding, W. Chen, et al., Identify origin of replication in saccharomyces cerevisiae using two-step feature selection technique, Bioinformatics, 35 (2019), 2075-2083. doi: 10.1093/bioinformatics/bty943
|
| [48] |
C. Q. Feng, Z. Y. Zhang, X. J. Zhu, Y. Lin, W. Chen, H. Tang, et al., iTerm-PseKNC: A sequence-based tool for predicting bacterial transcriptional terminators, Bioinformatics, 35 (2019), 1469-1477. doi: 10.1093/bioinformatics/bty827
|
| [49] | J. Shao, K. Yan, B. Liu, FoldRec-C2C: protein fold recognition by combining cluster-to-cluster model and protein similarity network, Brief Bioinform., (2020), DOI: 10.1093/bib/bbaa144. |
| [50] |
L. Cheng, Computational and biological methods for gene therapy, Curr. Gene Ther., 19 (2019), 210-210. doi: 10.2174/156652321904191022113307
|
| [51] |
L. Cheng, H. Zhao, P. Wang, W. Zhou, M. Luo, T. Li, et al., Computational methods for identifying similar diseases, Mol. Ther. Nucleic Acids., 18 (2019), 590-604. doi: 10.1016/j.omtn.2019.09.019
|
| [52] |
L. Cheng, C. Qi, H. Zhuang, T. Fu, X. Zhang, gutMDisorder: A comprehensive database for dysbiosis of the gut microbiota in disorders and interventions, Nucleic Acids Res., 48 (2020), D554-D560. doi: 10.1093/nar/gkz843
|
| [53] | H. Zulfiqar, M. S. Masoud, H. Yang, S. G. Han, C. Y. Wu, H. Lin, Screening of prospective plant compounds as H1R and CL1R inhibitors and its antiallergic efficacy through molecular docking approach, Comput. Math. Methods Med., 2021 (2021), 9. |
| [54] |
X. J. Zhu, C. Q. Feng, H. Y. Lai, W. Chen, L. Hao, Predicting protein structural classes for low-similarity sequences by evaluating different features, Knowl. Based Syst., 163 (2019), 787-793. doi: 10.1016/j.knosys.2018.10.007
|
| [55] |
Q. Zou, J. Zeng, L. Cao, R. Ji, A novel features ranking metric with application to scalable visual and bioinformatics data classification, Neurocomputing, 173 (2016), 346-354. doi: 10.1016/j.neucom.2014.12.123
|
| [56] | N. Rachburee, W. Punlumjeak, A comparison of feature selection approach between greedy, ig-ratio, chi-square, and mRMR in educational mining, in 2015 7th International Conference on Information Technology and Electrical Engineering (ICITEE), IEEE, (2015), 420-424. |
| [57] | Z. M. Zhang, J. S. Wang, H. Zulfiqar, H. Lv, F. Y. Dao, H. Lin, Early diagnosis of pancreatic ductal adenocarcinoma by combining relative expression orderings with machine learning method, Front. Cell Dev. Biol., 8 (2020), 1076. |
| [58] |
H. Peng, F. Long, C. Ding, Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy, IEEE Trans. Pattern Anal. Mach. Intell., 27 (2005), 1226-1238. doi: 10.1109/TPAMI.2005.159
|
| [59] |
J. X. Tan, S. H. Li, Z. M. Zhang, C. X. Chen, W. Chen, H. Tang, et al., Identification of hormone binding proteins based on machine learning methods, Math. Biosci. Eng., 16 (2019), 2466-2480. doi: 10.3934/mbe.2019123
|
| [60] |
H. Lv, Z. M. Zhang, S. H. Li, J. X. Tan, W. Chen, H. Lin, Evaluation of different computational methods on 5-methylcytosine sites identification, Brief Bioinform., 21 (2020), 982-995. doi: 10.1093/bib/bbz048
|
| [61] |
X. Li, L. Wang, E. Sung, AdaBoost with svm-based component classifiers, Eng. Appl. Artif. Intell., 21 (2008), 785-795. doi: 10.1016/j.engappai.2007.07.001
|
| [62] |
E. Frank, M. Hall, L. Trigg, G. Holmes, I. H. Witten, Data mining in bioinformatics using weka, Bioinformatics., 20 (2004), 2479-2481. doi: 10.1093/bioinformatics/bth261
|
| [63] |
X. Ru, L. Li, Q. Zou, Incorporating distance-based top-n-gram and random forest to identify electron transport proteins, J. Proteom. Res., 18 (2019), 2931-2939. doi: 10.1021/acs.jproteome.9b00250
|
| [64] |
Z. Lv, J. Zhang, H. Ding, Q. Zou, RF-PseU: A random forest predictor for RNA pseudouridine sites, Front. Bioeng. Biotechnol., 8 (2020), 134. doi: 10.3389/fbioe.2020.00134
|
| [65] | L. Breiman, Random forests, Mach Learn., 45 (2001), 5-32. |
| [66] | A. Abraham, F. Pedregosa, M. Eickenberg, P. Gervais, A. Mueller, J. Kossaifi, et al., Machine learning for neuroimaging with scikit-learn, Front. Neuroinform., 8 (2014), 14. |
| [67] |
P. Liang, W. Yang, X. Chen, C. Long, L. Zheng, H. Li, et al., Machine learning of single-cell transcriptome highly identifies mRNA signature by comparing f-score selection with DGE analysis, Mol. Ther. Nucleic Acids., 20 (2020), 155-163. doi: 10.1016/j.omtn.2020.02.004
|
| [68] | Z. D. Smith, A. Meissner, DNA methylation: Roles in mammalian development, Nat. Rev. Genet., 14 (2013), 204-220. |
| [69] |
K. Liu, W. Chen, H. Lin, XG-PseU: An extreme gradient boosting based method for identifying pseudouridine sites, Mol. Genet. Genom., 295 (2020), 13-21. doi: 10.1007/s00438-019-01600-9
|
| [70] |
V. Vacic, L. M. Iakoucheva, P. Radivojac, Two sample logo: a graphical representation of the differences between two sets of sequence alignments, Bioinformatics., 22 (2006), 1536-1537. doi: 10.1093/bioinformatics/btl151
|
| [71] |
Y. Zhang, Y. Li, R. Wang, J. Lu, X. Ma, M. Qiu, PSAC: Proactive sequence-aware content caching via deep learning at the network edge, IEEE Trans. Netw. Sci. Eng., 7 (2020), 2145-2154. doi: 10.1109/TNSE.2020.2990963
|
| [72] |
H. Lv, F. Y. Dao, D. Zhang, Z. X. Guan, H. Yang, W. Su, et al., iDNA-MS: An integrated computational tool for detecting DNA modification sites in multiple genomes, iScience, 23 (2020), 100991. doi: 10.1016/j.isci.2020.100991
|
| [73] | H. Xu, P. Jia, Z. Zhao, Deep4mC: Systematic assessment and computational prediction for DNA N4-methylcytosine sites by deep learning, Brief Bioinform., (2020), DOI: 10.1093/bib/bbaa099. |
| [74] | Q. Liu, J. Chen, Y. Wang, S. Li, C. Jia, J. song, et al., DeepTorrent: A deep learning-based approach for predicting DNA N4-methylcytosine sites, Brief Bioinform., (2020), DOI: 10.1093/bib/bbaa124. |
Figures(6) / Tables(5)

Hasan Zulfiqar, Rida Sarwar Khan, Farwa Hassan, Kyle Hippe, Cassandra Hunt, Hui Ding, Xiao-Ming Song, Renzhi Cao. Computational identification of N4-methylcytosine sites in the mouse genome with machine-learning method[J]. Mathematical Biosciences and Engineering, 2021, 18(4): 3348-3363. doi: 10.3934/mbe.2021167







 DownLoad:
DownLoad: