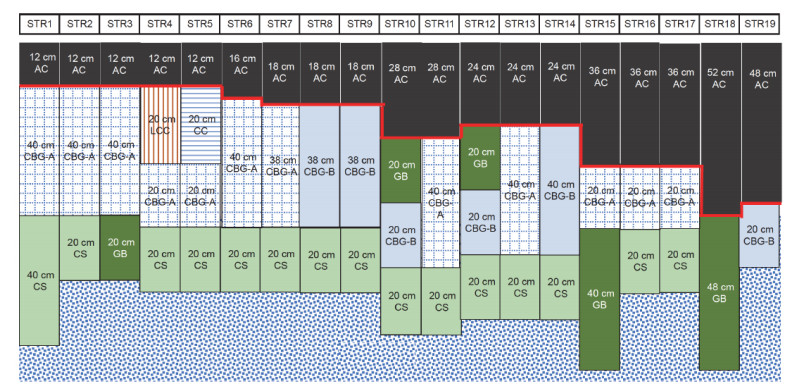
Semi-rigid asphalt pavement has a wide range of application cases and data bases, and rutting is a typical failure mode of semi-rigid asphalt pavement. The establishment of an accurate rutting depth prediction model is of great significance to pavement design and maintenance. However, due to the lack of perfect theoretical system and systematic research data, the existing rutting prediction model of semi-rigid asphalt pavement is not accurate. In this paper, machine learning and mechanical-empirical model are combined to study the feature selection affecting the rutting evolution and rutting depth model of semi-rigid asphalt pavement. First, the particle swarm optimization random forest model is used to select the important features that affect the evolution of rutting depth. Second, the R-F model based on important features is proposed for the first time, which is compared with modification of rutting model in the Chinese Specifications for Design of Highway Asphalt Pavement (JTG D50-2017) and R-B model based on the improved Burgers model. The results show that the R-F model has more accurate prediction ability and better generalization ability, and it does not need complex data preprocessing and noise reduction. Here, the machine learning method is introduced to analyze the data characteristics, and the R-F rutting depth prediction model framework is innovatively proposed, which greatly improves the applicability and accuracy of the existing model framework.
Citation: Bo Kou, Jinde Cao, Wei Huang, Tao Ma. The rutting model of semi-rigid asphalt pavement based on RIOHTRACK full-scale track[J]. Mathematical Biosciences and Engineering, 2023, 20(5): 8124-8145. doi: 10.3934/mbe.2023353
Semi-rigid asphalt pavement has a wide range of application cases and data bases, and rutting is a typical failure mode of semi-rigid asphalt pavement. The establishment of an accurate rutting depth prediction model is of great significance to pavement design and maintenance. However, due to the lack of perfect theoretical system and systematic research data, the existing rutting prediction model of semi-rigid asphalt pavement is not accurate. In this paper, machine learning and mechanical-empirical model are combined to study the feature selection affecting the rutting evolution and rutting depth model of semi-rigid asphalt pavement. First, the particle swarm optimization random forest model is used to select the important features that affect the evolution of rutting depth. Second, the R-F model based on important features is proposed for the first time, which is compared with modification of rutting model in the Chinese Specifications for Design of Highway Asphalt Pavement (JTG D50-2017) and R-B model based on the improved Burgers model. The results show that the R-F model has more accurate prediction ability and better generalization ability, and it does not need complex data preprocessing and noise reduction. Here, the machine learning method is introduced to analyze the data characteristics, and the R-F rutting depth prediction model framework is innovatively proposed, which greatly improves the applicability and accuracy of the existing model framework.

| [1] |
T. B. Moghaddam, M. R. Karim, M. A. Abdelaziz, A Review on fatigue and rutting performance of asphalt mixes, Sci. Res. Essays, 6 (2011), 670–682. https://doi.org/10.5897/SRE10.946 doi: 10.5897/SRE10.946

|
| [2] |
K. A. Ghuzlan, B. W. Al-Mistarehi, A. S. Al-Momani, Rutting performance of asphalt mixtures with gradations designed using Bailey and conventional Superpave methods, Constr. Build. Mater., 261 (2020), 119941. https://doi.org/10.1016/j.conbuildmat.2020.119941 doi: 10.1016/j.conbuildmat.2020.119941
|
| [3] |
L. L. Chen, G. Liu, Z. D. Qian, X. R. Zhang, Determination of allowable rutting depth based on driving safety analysis, J. Transp. Eng. Part B-Pavements, 146 (2020), 04020023. https://doi.org/10.1061/JPEODX.0000180 doi: 10.1061/JPEODX.0000180
|
| [4] |
L. Chen, G. Liu, B. Yao, Rutting prediction model for semi-rigid base asphalt pavement based on hamburg wheel tracking test, Int. J. Numer. Anal. Methods Geomech., 21 (2021), 1–8. https://doi.org/10.1061/(ASCE)GM.1943-5622.0002194 doi: 10.1061/(ASCE)GM.1943-5622.0002194
|
| [5] | R. D. Barksdale, Laboratory evaluation of rutting in base course materials, in Presented at the Third International Conference on the Structural Design of Asphalt Pavements, 1 (1972), 161–174. https://trid.trb.org/View/138844 |
| [6] | M. Venigalla, Modernization of commercial vehicle fleet on Indian roads, Master's Thesis Submitted to the Indian Institute of Technology, Madras, 1987. https://doi.org/10.13140/RG.2.2.14511.59046 |
| [7] |
L. du Plessis, N. F. Coetzee, T. P. Hoover, J. T. Harvey, Three decades of development and achievements: the heavy vehicle simulator in accelerated pavement testing, ASCE, (2006), 45–54. https://doi.org/10.1061/40866(198)7 doi: 10.1061/40866(198)7
|
| [8] |
H. J. Lee, Y. R. Kim, Viscoelastic constitutive model for asphalt concrete under cyclic loading, J. Eng. Mech. Div., Am. Soc. Civ. Eng., 124 (1998), 32–40. https://doi.org/10.1061/(ASCE)0733-9399(1998)124:1(32) doi: 10.1061/(ASCE)0733-9399(1998)124:1(32)
|
| [9] |
B. S. Underwood, Y. R. Kim, M. N. Guddati, Improved calculation method of damage parameter in viscoelastic continuum damage model, Int. J. Pavement Eng., 11 (2010), 459–476. https://doi.org/10.1080/10298430903398088 doi: 10.1080/10298430903398088
|
| [10] |
J. Zhang, L. L. Bao, Determination of asphalt mixture's viscoelastic constitutive parameters for pavement response analysis using dynamic modulus transformation, Constr. Build. Mater., 315 (2022), 1–12. https://doi.org/10.1016/j.conbuildmat.2021.125729 doi: 10.1016/j.conbuildmat.2021.125729
|
| [11] |
L. Tashman, E. Masad, D. Little, H. Zbib, A microstructure-based viscoplastic model for asphalt concrete, Int. J. Plast., 21 (2005), 1659–1685. https://doi.org/10.1016/j.ijplas.2004.11.008 doi: 10.1016/j.ijplas.2004.11.008
|
| [12] |
M. K. Darabi, R. K. A. Al-Rub, E. A. Masad, C. V. Huang, A thermo-viscoelastic-viscoplastic-viscodamage constitutive model for asphaltic materials, Int. J. Solids Struct., 48 (2011), 191–207. https://doi.org/10.1016/j.ijsolstr.2010.09.019 doi: 10.1016/j.ijsolstr.2010.09.019
|
| [13] |
L. L. Chen, G. Liu, Z. D. Qian, X. R. Zhang, Determination of allowable rutting depth based on driving safety analysis, J. Aerosp. Transp. Div., Am. Soc. Civ. Eng., 146 (2020), 04020023. https://doi.org/10.1061/JPEODX.0000180 doi: 10.1061/JPEODX.0000180
|
| [14] |
S. Hussan, M. A. Kamal, I. Hafeez, D. Farooq, N. Ahmad, S. Khanzada, Statistical evaluation of factors affecting the laboratory rutting susceptibility of asphalt mixtures, Int. J. Pavement Eng., 20 (2019), 402–416. https://doi.org/10.1080/10298436.2017.1299527 doi: 10.1080/10298436.2017.1299527
|
| [15] |
V. Radhakrishnan, R. R. Dudipala, A. Maity, K. S. Reddy, Evaluation of rutting potential of asphalts using resilient modulus test parameters, Road Mater. Pavement Des., 20 (2019), 20–35. https://doi.org/10.1080/14680629.2017.1374994 doi: 10.1080/14680629.2017.1374994
|
| [16] |
S. Hussan, M. A. Kamal, I. Hafeez, N. Ahmad, S. Khanzada, S. Ahmed, Modelling asphalt pavement analyzer rut depth using different statistical techniques, Road Mater. Pavement Des., 21 (2020), 117–142. https://doi.org/10.1080/14680629.2018.1481880 doi: 10.1080/14680629.2018.1481880
|
| [17] |
P. Singh, A. K. Swamy, Probabilistic approach to characterise laboratory rutting behaviour of asphalt concrete mixtures, Int. J. Pavement Eng., 21 (2018), 384–396. https://doi.org/10.1080/10298436.2018.1480780 doi: 10.1080/10298436.2018.1480780
|
| [18] |
A. K. Singh, J. P. Sahoo, Rutting prediction models for flexible pavement structures: A review of historical and recent developments, J. Aerosp. Transp. Div., Am. Soc. Civ. Eng., 8 (2021), 315–338. https://doi.org/10.1016/j.jtte.2021.04.003 doi: 10.1016/j.jtte.2021.04.003
|
| [19] |
J. F. Shook, Construction materials control–AASHO road test, J. Soil Mech. Found. Div., 85 (1959), 15–29. https://doi.org/10.1061/JSFEAQ.0000219 doi: 10.1061/JSFEAQ.0000219
|
| [20] |
C. Petit, E. Manyo, P. Reynaud, The French research project on roadway lifespan design of supface layers and on-site measurement methods, Rev. Gen. Des. Routes, 952 (2018), 42–45. https://doi.org/10.1002/eqe.685 doi: 10.1002/eqe.685
|
| [21] |
M. S. Mamlouk, J. O. Cane, Implementation of the 1986 aashto guide at the city and counn levels, Technol. Forecast. Soc. Change, 1989. https://doi.org/10.1016/j.techfore.2004.07.003 doi: 10.1016/j.techfore.2004.07.003
|
| [22] |
X. D. Wang, G. L. Zhou, H. Y. Liu, Q. Xiao, Key points of RIOHTRACK testing road design and construction, J. Highw. Transp. Res. Dev., 14 (2020), 1–16. https://doi.org/10.1061/JHTRCQ.0000749 doi: 10.1061/JHTRCQ.0000749
|
| [23] | J. B. Metcalf, A brief history of full-scale accelerated pavement testing facilities to 1962, in The Roles of Accelerated Pavement Testing in Pavement Sustainability, Springer International Publishing Switzerland, 2016. https://doi.org/10.1007/978-3-319-42797-3_1 |
| [24] |
L. Zhang, X. Y. Zhou, X. D. Wang, Research progress of long-life asphalt pavement behavior based on the RIOHTRACK full-scale accelerated loading test, Chin. J., 65 (2020), 3247–3258. https://doi.org/10.1360/TB-2020-0287 doi: 10.1360/TB-2020-0287
|
| [25] |
Y. L. Pavlov, Random Forests, Karelian Centre Russian Acad. Sci. Petrozavodsk, 45 (1997), 5–32. https://doi.org/10.1023/A:1010933404324 doi: 10.1023/A:1010933404324
|
| [26] |
T. Dietterich, Ensemble methods in machinelearning, Lect. Notes Comput. Sci., 1857 (2000), 1–15. https://doi.org/10.1007/3-540-45014-9_1 doi: 10.1007/3-540-45014-9_1
|
| [27] |
B. H. Menze, B. M. Kelm, R. Masuch, U. Himmelreich, P. Bachert, W. Petrich, et al., A comparison of random forest and its gini importance with standard chemometric methods for the feature selection and classification of spectral data, BMC Bioinf., 10 (2009), 213. https://doi.org/10.1186/1471-2105-10-213 doi: 10.1186/1471-2105-10-213
|
| [28] |
K. J. Archer, R. V. Kimes, Empirical characterization of random forest variable importance measures, Comput. Stat. Data An., 52 (2008), 2249–2260. https://doi.org/10.1016/j.csda.2007.08.015 doi: 10.1016/j.csda.2007.08.015
|
| [29] |
Y. D. Zhang, S. H. Wang, P. Phillips, G. Ji, Binary PSO with mutation operator for feature selection using decision tree applied to spam detection, Knowl.-Based Syst., 64 (2014), 22–31. https://doi.org/10.1016/j.knosys.2014.03.015 doi: 10.1016/j.knosys.2014.03.015
|
| [30] |
V. Haritonovs, J. Smirnovs, J. Nauduns, Prediction of rutting formation in asphalt concrete pavement, Balt. J. Road Bridge Eng., 5 (2010), 38–42. https://doi.org/10.3846/BJRBE.2010.05 doi: 10.3846/BJRBE.2010.05
|
| [31] |
N. Saboo, A. Mudgal, Modelling creep and recovery response of asphalt binders using generalized burgers model, J. Pet. Sci. Technol., 36 (2018), 1627–1634. https://doi.org/10.1080/10916466.2018.1496109 doi: 10.1080/10916466.2018.1496109
|
| [32] |
L. Gao, H. Dan, J. Chen, Research on predicting the rutting of asphalt pavement based on a simplified burgers creep model, Math. Probl. Eng., 2017 (2017), 3459704. https://doi.org/10.1155/2017/3459704 doi: 10.1155/2017/3459704
|
| [33] |
H. Behbahani, H. Ziari, N. Kamboozia, Evaluation of the visco-elasto-plastic behavior of glasphalt mixtures through generalized and classic Burger's models modification, Constr. Build. Mater., 118 (2016), 36–42. https://doi.org/10.1016/j.conbuildmat.2016.04.157 doi: 10.1016/j.conbuildmat.2016.04.157
|
| [34] |
B. Dokku, D. Savio, M. R. Nivitha, J. M. Krishnan, Development of rutting model for Indian highways based on rut depth simulations from AASHTO ware pavement ME design software, J. Transp. Eng. Pt. B-Pavements, 146 (2020), 04020023. https://doi.org/10.1061/JPEODX.0000160 doi: 10.1061/JPEODX.0000160
|
| [35] |
N. Hossain, D. Singh, M. Zaman, Enhancing rutting prediction of the mechanistic-empirical pavement design guide by using data from a field test section in Oklahoma, Transp. Res. Record, 2590 (2016), 28–36. https://doi.org/10.3141/2590-04 doi: 10.3141/2590-04
|








Figures(9) / Tables(2)

Bo Kou, Jinde Cao, Wei Huang, Tao Ma. The rutting model of semi-rigid asphalt pavement based on RIOHTRACK full-scale track[J]. Mathematical Biosciences and Engineering, 2023, 20(5): 8124-8145. doi: 10.3934/mbe.2023353







 DownLoad:
DownLoad: