This study aimed to analyze the potential genes associated with immune cell infiltration in atherosclerosis (AS).
Gene expression profile data (GSE57691) of human arterial tissue samples were downloaded, and differentially expressed RNAs (DERNAs; long-noncoding RNA [lncRNAs], microRNAs [miRNAs], and messenger RNAs [mRNAs]) in AS vs. control groups were selected. Based on genome-wide expression levels, the proportion of infiltrating immune cells in each sample was assessed. Genes associated with immune infiltration were selected, and subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. Finally, a competing endogenous RNA (ceRNA) network was constructed, and the genes in the network were subjected to functional analyses.
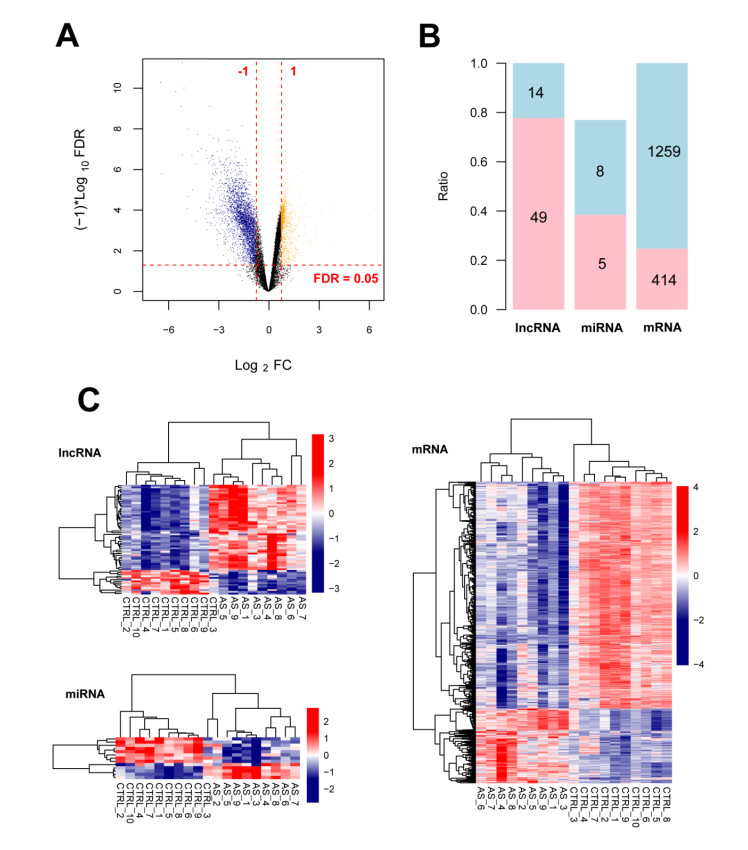
A total of 1749 DERNAs meeting the thresholds were screened, including 1673 DEmRNAs, 63 DElncRNAs, and 13 DEmiRNAs. The proportions of B cells, CD4+ T cells, and CD8+ T cells were significantly different between the two groups. In total, 341 immune-associated genes such as HBB, FCN1, IL1B, CXCL8, RPS27A, CCN3, CTSZ, and SERPINA3 were obtained that were enriched in 70 significantly related GO biological processes (such as immune response) and 15 KEGG pathways (such as chemokine signaling pathway). A ceRNA network, including 33 lncRNAs, 11 miRNAs, and 216 mRNAs, was established.
Genes such as FCN1, IL1B, and SERPINA3 may be involved in immune cell infiltration and may play important roles in AS progression via ceRNA regulation.
Citation: Xiaodong Xia, Manman Wang, Jiao Li, Qiang Chen, Heng Jin, Xue Liang, Lijun Wang. Identification of potential genes associated with immune cell infiltration in atherosclerosis[J]. Mathematical Biosciences and Engineering, 2021, 18(3): 2230-2242. doi: 10.3934/mbe.2021112
This study aimed to analyze the potential genes associated with immune cell infiltration in atherosclerosis (AS).
Gene expression profile data (GSE57691) of human arterial tissue samples were downloaded, and differentially expressed RNAs (DERNAs; long-noncoding RNA [lncRNAs], microRNAs [miRNAs], and messenger RNAs [mRNAs]) in AS vs. control groups were selected. Based on genome-wide expression levels, the proportion of infiltrating immune cells in each sample was assessed. Genes associated with immune infiltration were selected, and subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. Finally, a competing endogenous RNA (ceRNA) network was constructed, and the genes in the network were subjected to functional analyses.
A total of 1749 DERNAs meeting the thresholds were screened, including 1673 DEmRNAs, 63 DElncRNAs, and 13 DEmiRNAs. The proportions of B cells, CD4+ T cells, and CD8+ T cells were significantly different between the two groups. In total, 341 immune-associated genes such as HBB, FCN1, IL1B, CXCL8, RPS27A, CCN3, CTSZ, and SERPINA3 were obtained that were enriched in 70 significantly related GO biological processes (such as immune response) and 15 KEGG pathways (such as chemokine signaling pathway). A ceRNA network, including 33 lncRNAs, 11 miRNAs, and 216 mRNAs, was established.
Genes such as FCN1, IL1B, and SERPINA3 may be involved in immune cell infiltration and may play important roles in AS progression via ceRNA regulation.

| [1] | A. Gistera, G. K. Hansson, The immunology of atherosclerosis, Nat. Rev. Nephrol., 13 (2017), 368-380. |
| [2] | I. Gyárfás, M. Keltai, Y. Salim, Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the interheart study): Case-control study, Orvosi. Hetil., 147 (2006), 675-686. |
| [3] |
M. Nus, Z. Mallat, Immune-mediated mechanisms of atherosclerosis and implications for the clinic, Expert Rev. Clin. Immunol., 12 (2016), 1217-1237. doi: 10.1080/1744666X.2016.1195686

|
| [4] |
L. Saba, T. Saam, H. R. Jäger, C. Yuan, T. S. Hatsukami, D. Saloner, et al., Imaging biomarkers of vulnerable carotid plaques for stroke risk prediction and their potential clinical implications, Lancet Neurol., 18 (2019), 559-572. doi: 10.1016/S1474-4422(19)30035-3
|
| [5] |
D. Baptista, F. Mach, K. J. Brandt, Follicular regulatory T cell in atherosclerosis, J. Leukoc. Biol., 104 (2018), 925-930. doi: 10.1002/JLB.MR1117-469R
|
| [6] | T. Shimokama, S. Haraoka, T. Watanabe, Immunohistochemical and ultrastructural demonstration of the lymphocyte-macrophage interaction in human aortic intima, Mod. pathol. Offic. J. United States Canadian Acad. Pathol., 4 (1991), 101-107. |
| [7] |
A. Hermansson, D. F. Ketelhuth, D. Strodthoff, M. Wurm, E. M. Hansson, A. Nicoletti, et al., Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis, J. Exp. Med., 207 (2010), 1081-1093. doi: 10.1084/jem.20092243
|
| [8] |
D. Tsiantoulas, A. P. Sage, Z. Mallat, C. J. Binder, Targeting B cells in atherosclerosis: closing the gap from bench to bedside, Arterioscler, Thromb., Vasc. Biol., 35 (2015), 296-302. doi: 10.1161/ATVBAHA.114.303569
|
| [9] |
J. Xu, Y. Yang, Potential genes and pathways along with immune cells infiltration in the progression of atherosclerosis identified via microarray gene expression dataset re-analysis, Vascular, 28 (2020), 643-654. doi: 10.1177/1708538120922700
|
| [10] |
E. Biros, G. Gabel, C. S. Moran, C. Schreurs, J. H. N. Lindeman, P. J. Walker, et al., Differential gene expression in human abdominal aortic aneurysm and aortic occlusive disease, Oncotarget, 6 (2015), 12984-12996. doi: 10.18632/oncotarget.3848
|
| [11] |
T. Barrett, S. E. Wilhite, P. Ledoux, C. Evangelista, I. F. Kim, M. Tomashevsky, et al., NCBI GEO: Archive for functional genomics data sets-update, Nucleic Acids Res., 41 (2012), 991-995. doi: 10.1093/nar/gks1193
|
| [12] |
M. W. Wright, A short guide to long non-coding RNA gene nomenclature, Hum. Genomics, 8 (2014), 7. doi: 10.1186/1479-7364-8-7
|
| [13] |
M. E. Ritchie, B. Phipson, D. Wu, Y. Hu, C. W. Law, W. Shi, et al., Limma powers differential expression analyses for RNA-sequencing and microarray studies, Nucleic Acids Res., 43 (2015), e47. doi: 10.1093/nar/gkv007
|
| [14] |
L. Wang, C. Cao, Q. Ma, Q. Zeng, H. Wang, Z. Cheng, et al., RNA-seq analyses of multiple meristems of soybean: novel and alternative transcripts, evolutionary and functional implications, BMC Plant Biol., 14 (2014), 169-169. doi: 10.1186/1471-2229-14-169
|
| [15] | J. Racle, K. De Jonge, P. Baumgaertner, D. E. Speiser, D. Gfeller, Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data, eLife, 13 (2017), e26476. |
| [16] |
D. Huang, B. T. Sherman, R. A. Lempicki, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources, Nat. Protoc., 4 (2009), 44-57. doi: 10.1038/nprot.2008.211
|
| [17] |
L. Salmena, L. Poliseno, Y. Tay, L. Kats, P. P. Pandolfi, A ceRNA hypothesis: The rosetta stone of a hidden rna language?, Cell, 146 (2011), 353-358. doi: 10.1016/j.cell.2011.07.014
|
| [18] |
M. D. Paraskevopoulou, I. S. Vlachos, D. Karagkouni, G. Georgakilas, I. Kanellos, T. Vergoulis, et al., DIANA-LncBase v2: Indexing microRNA targets on non-coding transcripts, Nucleic Acids Res., 44 (2016), 231-238. doi: 10.1093/nar/gkv1270
|
| [19] | J. Li, S. Liu, H. Zhou, L. Qu, J. Yang, StarBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data, Nucleic Acids Res., 42 (2014), 92-97. |
| [20] |
P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, et al., Cytoscape: A software environment for integrated models of biomolecular interaction networks, Genome Res., 13 (2003), 2498-2504. doi: 10.1101/gr.1239303
|
| [21] |
V. Vianahuete, J. J. Fuster, Potential therapeutic value of interleukin 1b-targeted strategies in atherosclerotic cardiovascular disease, Rev. Esp. Cardiol., 72 (2019), 760-766. doi: 10.1016/j.recesp.2019.02.021
|
| [22] |
R. Zorcpleskovic, A. Pleskovic, O. Vraspirporenta, M. Zorc, A. Milutinovic, Immune cells and vasa vasorum in the tunica media of atherosclerotic coronary arteries, Bosn. J. Basic Med. Sci., 18 (2018), 240-245. doi: 10.17305/bjbms.2018.2951
|
| [23] |
D. A. Chistiakov, A. N. Orekhov, Y. V. Bobryshev, Immune-inflammatory responses in atherosclerosis: Role of an adaptive immunity mainly driven by T and B cells, Immunobiology, 221 (2016), 1014-1033. doi: 10.1016/j.imbio.2016.05.010
|
| [24] | X. Zhou, S. Stemme, G. K. Hansson, Evidence for a local immune response in atherosclerosis, CD4+ T cells infiltrate lesions of apolipoprotein-E-deficient mice, Am. J. pathol., 149 (1996), 359. |
| [25] |
C. Cochain, M. Koch, S. M. Chaudhari, M. Busch, J. Pelisek, L. Boon, et al., CD8+ T cells regulate monopoiesis and circulating Ly6C-high monocyte levels in atherosclerosis in mice, Circ. Res., 117 (2015), 244-253. doi: 10.1161/CIRCRESAHA.117.304611
|
| [26] |
T. Kimura, K. Tse, A. Sette, K. Ley, Vaccination to modulate atherosclerosis, Autoimmunity, 48 (2015), 152-160. doi: 10.3109/08916934.2014.1003641
|
| [27] |
M. Katayama, K. Ota, N. Nagimiura, N. Ohno, N. Yabuta, H. Nojima, et al., Ficolin-1 is a promising therapeutic target for autoimmune diseases, Int. Immunol., 31 (2019), 23-32. doi: 10.1093/intimm/dxy056
|
| [28] |
S. J. Catarino, F. A. Andrade, A. B. W. Boldt, L. Guilherme, I. J. Messias-Reason, Sickening or healing the heart? The association of ficolin-1 and rheumatic fever, Front. Immunol., 9 (2018), 3009. doi: 10.3389/fimmu.2018.03009
|
| [29] |
P. Libby, Interleukin-1 beta as a target for atherosclerosis therapy: Biological basis of cantos and beyond, J. Am. Coll. Cardiol., 70 (2017), 2278-2289. doi: 10.1016/j.jacc.2017.09.028
|
| [30] |
M. R. Alexander, M. Murgai, C. W. Moehle, G. K. Owens, Interleukin-1β modulates smooth muscle cell phenotype to a distinct inflammatory state relative to PDGF-DD via NF-κB-dependent mechanisms, Physiol. Genom., 44 (2012), 417-429. doi: 10.1152/physiolgenomics.00160.2011
|
| [31] |
V. Sorokin, C. C. Woo, Role of Serpina3 in vascular biology, Int. J. Cardiol., 304 (2020), 154-155. doi: 10.1016/j.ijcard.2019.12.030
|
| [32] |
L. Zhao, M. Zheng, Z. Guo, K. Li, Y. Liu, M. Chen, et al., Circulating Serpina3 levels predict the major adverse cardiac events in patients with myocardial infarction, Int. J. Cardiol., 300 (2020), 34-38. doi: 10.1016/j.ijcard.2019.08.034
|
| [33] |
D. Wagsater, D. X. Johansson, V. Fontaine, E. Vorkapic, A. Backlund, A. Razuvaev, et al., Serine protease inhibitor A3 in atherosclerosis and aneurysm disease, Int. J. Mol. Med., 30 (2012), 288-294. doi: 10.3892/ijmm.2012.994
|
| [34] |
A. J. Horvath, J. A. Irving, J. Rossjohn, R. H. Law, S. P. Bottomley, N. S. Quinsey, et al., The murine orthologue of human antichymotrypsin: a structural paradigm for clade A3 serpins, J. Biol. Chem., 280 (2005), 43168-43178. doi: 10.1074/jbc.M505598200
|





Figures(6)

Xiaodong Xia, Manman Wang, Jiao Li, Qiang Chen, Heng Jin, Xue Liang, Lijun Wang. Identification of potential genes associated with immune cell infiltration in atherosclerosis[J]. Mathematical Biosciences and Engineering, 2021, 18(3): 2230-2242. doi: 10.3934/mbe.2021112







 DownLoad:
DownLoad: