Citation: Conrad Ratchford, Jin Wang. Multi-scale modeling of cholera dynamics in a spatially heterogeneous environment[J]. Mathematical Biosciences and Engineering, 2020, 17(2): 948-974. doi: 10.3934/mbe.2020051

| [1] | B. Boldin and O. Diekmann, Superinfections can induce evolutionarily stable coexistence of pathogens, J. Math. Biol., 56 (2008), 635-672. |
| [2] | M. A. Gilchrist and A. Sasaki, Modeling host-parasite coevolution: a nested approach based on mechanistic models, J. Theor. Biol., 218 (2002), 289-308. |
| [3] | M. Marcheva, N. Tuncer and C. M. St. Mary, Coupling within-host and between-host infectious disease models, Biomath., 4 (2015), 1510091. |
| [4] | M. Martcheva and X. Z. Li, Linking immunological and epidemiological dynamics of HIV: the case of super-infection, J. Biol. Dynam., 7 (2013), 161-182. |
| [5] | N. Mideo, S. Alizon and T. Day, Linking within- and between-host disease dynamics, Trends Ecol. Evol., 23 (2008), 511-517. |
| [6] | M. Ali, A. R. Nelson, A. L. Lopez, et al., Updated global burden of cholera in endemic countries, PLoS Neglect. Trop. D., 9 (2015), e0003832. |
| [7] | WHO, Cholera Fact Sheet number 107: December 2017. Available from: http://www.who.int/mediacentre/factsheets/fs107/en/ |
| [8] | D. M. Hartley, J. G. Morris and D. L. Smith, Hyperinfectivity: a critical element in the ability of V. cholerae to cause epidemics? PLoS Med., 3 (2006), 0063-0069. |
| [9] | E. J. Nelson, J. B. Harris, J. G. Morris, et al., Cholera transmission: The host, pathogen and bacteriophage dynamics, Nat. Rev. Microbiol., 7 (2009), 693-702. |
| [10] | C. T. Codeço, Endemic and epidemic dynamics of cholera: the role of the aquatic reservoir, BMC Infect. Dis., 1 (2001), 1. |
| [11] | J. R. Andrews and S. Basu, Transmission dynamics and control of cholera in Haiti: an epidemic model, Lancet, 377 (2011), 1248-1255. |
| [12] |
Z. Mukandavire, S. Liao, J. Wang, et al., Estimating the reproductive numbers for the 2008-2009 cholera outbreaks in Zimbabwe, P. Natl. Acad. Sci. U.S.A., 108 (2011), 8767-8772. doi: 10.1073/pnas.1019712108

|
| [13] | L. Righetto, E. Bertuzzo, R. Casagrandi, et al., Modeling human movement in a cholera spreading along fluvial systems, Ecohydrol., 4 (2011), 49-55. |
| [14] | Z. Shuai and P. van den Driessche, Global dynamics of cholera models with differential infectivity, Math. Biosci., 234 (2011), 118-126. |
| [15] | J. P. Tian and J. Wang, Global stability for cholera epidemic models, Math. Biosci., 232 (2011), 31-41. |
| [16] | J. H. Tien and D. J. D. Earn, Multiple transmission pathways and disease dynamics in a waterborne pathogen model, Bull. Math. Biol., 72 (2010), 1506-1533. |
| [17] | A. R. Tuite, J. H. Tien, M. C. Eisenberg, et al., Cholera epidemic in Haiti, 2010: Using a transmission model to explain spatial spread of disease and identify optimal control interventions, Ann. Int. Med., 154 (2011), 293-302. |
| [18] | E. Bertuzzo, R. Casagrandi, M. Gatto, et al., On spatially explicit models of cholera epidemics, J. Royal Soc. Interface, 7 (2010), 321-333. |
| [19] | A. Rinaldo, E. Bertuzzo, L. Mari, et al., Reassessment of the 2010-2011 Haiti cholera outbreak and rainfall-driven multiseason projections, P. Natl. Acad. Sci. U.S.A., 109 (2012), 6602-6607. |
| [20] | D. He, X. Wang, D. Gao, et al., Modeling the 2016-2017 Yemen cholera outbreak with the impact of limited medical resources, J. Theor. Biol., 451 (2018), 80-85. |
| [21] | X. Wang and J. Wang, Analysis of cholera epidemics with bacterial growth and spatial movement, J. Biol. Dynam., 9 (2015), 233-261. |
| [22] | X. Wang, D. Posny and J. Wang, A Reaction-Convection-Diffusion Model for Cholera Spatial Dynamics, Discrete Cont. Dyn-S, 21 (2016), 2785-2809. |
| [23] | X. Wang, X.-Q. Zhao and J. Wang, A cholera epidemic model in a spatiotemporally heterogeneous environment, J. Math. Anal. Appl., 468 (2018), 893-912. |
| [24] | E. Bertuzzo, L. Mari, L. Righetto, et al., Prediction of the spatial evolution and effects of control measures for the unfolding Haiti cholera outbreak, Geophys. Res. Lett., 38 (2011), L06403. |
| [25] | M. C. Eisenberg, Z. Shuaic, J. H. Tien, et al., A cholera model in a patchy environment with water and human movement, Math. Biosci., 246 (2013), 105-112. |
| [26] | M. K. Waldor and J. J. Mekalanos, Lysogenic conversion by a filamentous phage encoding cholera toxin, Science, 272 (1996), 1910-1914. |
| [27] | R. R. Colwell, A global and historical perspective of the genus Vibrio, in The Biology of Vibrios, F.L. Thompson, B. Austin, and J. Swings (eds.), ASM Press, Washington DC, 2006. |
| [28] | X. Wang and J. Wang, Disease dynamics in a coupled cholera model linking within-host and between-host interactions, J. Biol. Dynam., 11 (2017), 238-262. |
| [29] | X. Wang and J. Wang, Modeling the within-host dynamics of cholera: bacterial-viral interaction, J. Biol. Dynam., 11 (2017), 484-501. |
| [30] | C. Ratchford and J. Wang, Modeling cholera dynamics at multiple scales: environmental evolution, between-host transmission, and within-host interaction, Math. Biosci. Eng., 16 (2019), 782-812. |
| [31] | H. Guo, M. Li and Z. Shuai, Global stability of the endemic equilibrium of multi-group SIR epidemic models, Can. Appl. Math. Q., 14 (2006), 259-283. |
| [32] | R. Sun and J. Shi, Global stability of multigroup epidemic model with group mixing and nonlinear incidence rates, Appl. Math. Comput., 218 (2011), 280-286. |
| [33] | C. Cosner, J. C. Beier, R. S. Cantrell, et al., The effects of human movement on the persistence of vector-borne diseases, J. Theor. Biol., 258 (2009), 550-560. |
| [34] | Z. Shuai and P. van den Driessche, Modeling and control of cholera on networks with a common water source, J. Biol. Dynam., 9 (2015), 90-103. |
| [35] | P. van den Driessche and J. Watmough, Reproduction numbers and sub-threshold endemic equilibria for compartmental models of disease transmission, Math. Biosci., 180 (2002), 29-48. |
| [36] | Z. Shuai and P. van den Driessche, Global stability of infectious disease models using Lyapunov functions, SIAM J. Appl. Math., 73 (2013), 1513-1532. |
| [37] | M. Li and Z. Shuai, Global-stability problem for coupled systems of differential equations on networks, J. Differ. Equations, 248 (2010), 1-20. |
| [38] | N. M. Ferguson, D. A. T. Cummings, S. Cauchemez, et al., Strategies for containing an emerging influenza pandemic in Southeast Asia, Nature, 437 (2005), 209-214. |
| [39] | B. Roche, J. M. Drake and P. Rohani, An agent-based model to study the epidemiological and evolutionary dynamics of Influenza viruses, BMC Bioinform., 12 (2011), 87. |
| [40] | E. Bonabeau, Agent-based modeling: Methods and techniques for simulating human systems, P. Natl. Acad. Sci. U.S.A., 99 (2002), 7280-7287. |
| [41] | P. Kumberger, K. Durso-Cain, S. Uprichard, et al., Accounting for space - Quantification of cellto-cell transmission kinetics using virus dynamics models, Viruses, 10 (2018), 200. |
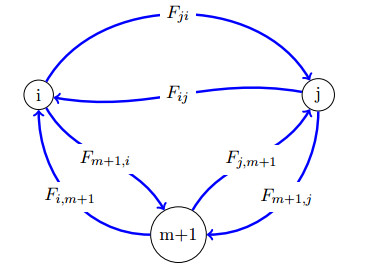
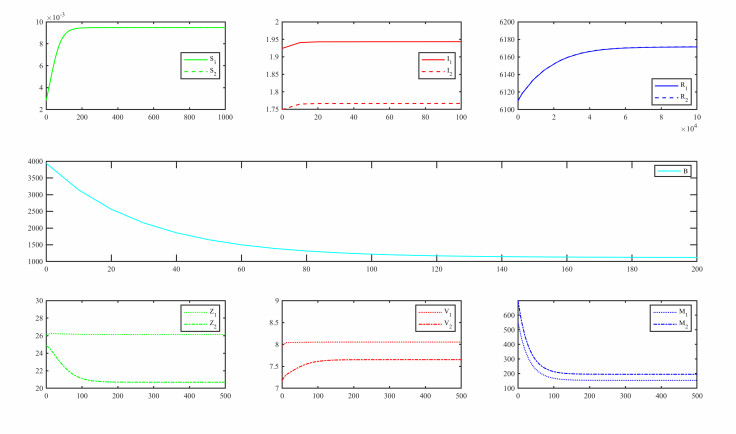

Figures(3)

Conrad Ratchford, Jin Wang. Multi-scale modeling of cholera dynamics in a spatially heterogeneous environment[J]. Mathematical Biosciences and Engineering, 2020, 17(2): 948-974. doi: 10.3934/mbe.2020051







 DownLoad:
DownLoad: